




2011-2017 © МБУЗ ГКП № 7, г.Челябинск.
Гемоглобин входит в состав группы белков гемопротеины, которые входят в класс хромопротеинов, и подразделяются на неферментативные белки (гемоглобин, миоглобин) и ферменты (цитохромы, каталаза, пероксидаза). Небелковой частью их является гем – структура, включающая в себя порфириновое кольцо (состоящее из 4 пиррольных колец) и иона Fe2+. Железо связывается с порфириновым кольцом двумя координационными и двумя ковалентными связями.
Гемоглобин представляет собой белок, включающий 4 гемсодержащие белковые субъединицы. Между собой протомеры соединяются гидрофобными, ионными, водородными связями, при этом они взаимодействуют не произвольно, а определенным участком – контактной поверхностью. Этот процесс высокоспецифичен, контакт происходит одновременно в десятках точек
по принципу комплементарности. Взаимодействие осуществляют разноименно заряженные группы, гидрофобные участки, неровности на поверхности белка.
Белковые субъединицы в нормальном гемоглобине могут быть представлены различными типами полипептидных цепей: α, β, γ, δ, ε, ξ (соответственно, греч. - альфа, бета, гамма, дельта, эпсилон, кси). В состав молекулы гемоглобина входят по две цепи двух разных типов.
Гем соединяется с белковой субъединицей, во-первых, через остаток гистидина координационной связью железа, во-вторых, через гидрофобные связи пиррольных колец и гидрофобных аминокислот. Гем располагается как бы "в кармане" своей цепи и формируется гемсодержащий протомер.
Существует несколько нормальных вариантов гемоглобина:

HbS – гемоглобин серповидно-клеточной анемии.
MetHb – метгемоглобин, форма гемоглобина, включающая трехвалентный ион железа вместо двухвалентного. Такая форма образуется спонтанно, при взаимодействии молекулы O2 и гемового Fe2+, но обычно ферментативных мощностей клетки хватает на его восстановление (НАДН- и НАДФН-зависимые метгемоглобин-редуктазы и глутатионовая антиоксидантная система).
При использовании сульфаниламидов, употреблении нитрита натрия и нитратов пищевых продуктов, при недостаточности аскорбиновой кислоты ускоряется переход Fe 2+ в Fe3+. Образующийся metHb не способен связывать кислород и возникает гипоксия тканей. Для восстановления Fe3+ в Fe2+ в клинике используют аскорбиновую кислоту и метиленовую синь.
Hb-CO – карбоксигемоглобин, образуется при наличии СО (угарный газ) во вдыхаемом воздухе. Он постоянно присутствует в крови в малых концентрациях, но его доля может колебаться от условий и образа жизни.
Угарный газ является активным ингибитором гем-содержащих ферментов, в частности, цитохромоксидазы, 4-го комплекса дыхательной цепи.
Карбоксигемоглобин присутствует и в норме в количестве 0,5-1,5%, в сельской местности меньше, чем в городе. У курильщиков концентрация Hb-CO возрастает, в зависимости от количества сигарет в день, до 8-9%.
HbA1С – гликозилированный гемоглобин. Концентрация его нарастает при хронической гипергликемии и является хорошим скрининговым показателем уровня глюкозы крови за длительный период времени (время жизни эритроцита, 3-4 месяца).
Французские и американские ученые сообщили о первом случае излечения человека от серповидноклеточной анемии с помощью генной терапии. Пациентом стал подросток из Франции. Описание случая приводится в New England Journal of Medicine.
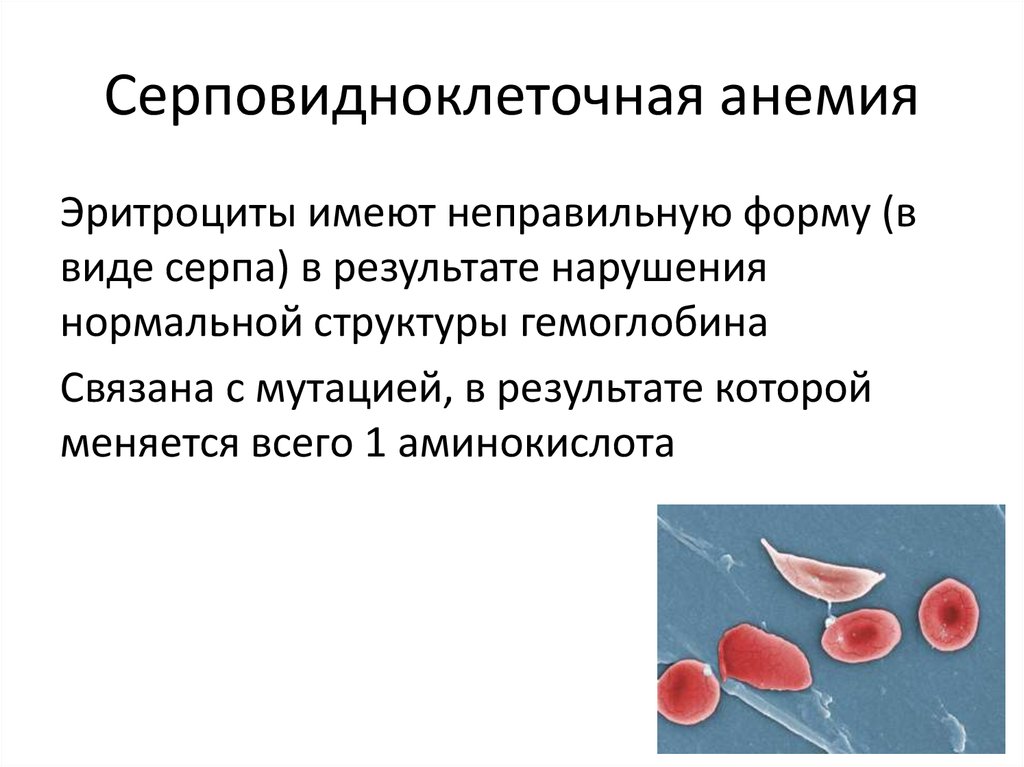
Серповидноклеточная анемия — это наследственное заболевание, обусловленное одиночной мутацией в гене бета-субъединицы гемоглобина (белковая часть гемоглобина состоит из двух альфа- и двух бета-субъединиц). Дефектный гемоглобин (HbS), отдавая тканям кислород, полимеризуется с образованием волокон, которые деформируют эритроциты, придавая им серповидную форму. Такие эритроциты хуже проходят через мелкие сосуды, что приводит к образованию тромбов, а также быстрее разрушаются, из-за чего возникает анемия.
Дефектный гемоглобин (HbS), отдавая тканям кислород, полимеризуется с образованием волокон, которые деформируют эритроциты, придавая им серповидную форму. Такие эритроциты хуже проходят через мелкие сосуды, что приводит к образованию тромбов, а также быстрее разрушаются, из-за чего возникает анемия.
Для лечения серповидноклеточной анемии в настоящее время одобрено только одно лекарство — гидроксимочевина. Она способствует синтезу младенческой (фетальной) формы гемоглобина, которая снижает концентрацию HbS и замедляет его полимеризацию. Однако препарат действует не на все клетки и обладает множеством побочных эффектов. Небольшому числу пациентов (примерно 18 процентам) можно помочь пересадкой донорского костного мозга. Подобные виды лечения не могут покрыть потребности всех больных, и ученые на протяжении десятков лет занимались разработкой генной терапии заболевания. После успешных ее испытаний на животных в 2014 году начались клинические испытания этого метода лечения.
Одним из участников испытаний, которые продолжаются до сих пор на базе Детской больнице Неккер в Париже, стал мальчик, болевший серповидноклеточной анемией с рождения и наблюдавшийся в этом медучреждении. В возрасте от двух до девяти лет пациент получал гидроксимочевину без выраженного эффекта: у него наблюдались повторные тромбозы мелких сосудов (в том числе дважды — легочных) и двусторонний очаговый некроз бедренных костей. С 2010 года ему проводили профилактические переливания эритроцитарной массы.
В возрасте от двух до девяти лет пациент получал гидроксимочевину без выраженного эффекта: у него наблюдались повторные тромбозы мелких сосудов (в том числе дважды — легочных) и двусторонний очаговый некроз бедренных костей. С 2010 года ему проводили профилактические переливания эритроцитарной массы.
В октябре 2014 года, когда мальчику было 13 лет, ему провели генную терапию в соответствии с протоколом, разработанным французскими учеными и американской компанией Bluebird Bio. На первом этапе лечения у пациента дважды забрали ткань костного мозга (для проведения терапии и создания резервного запаса) и выделили из нее мультипотентные кроветворные клетки (CD34+). В эти клетки с помощью обезвреженного лентивируса ввели ген здорового варианта бета-глобина βA-T87Q. Средняя доза геннотерапевтического препарата LentiGlobin BB305 составила 1,0 и 1,2 копии вирусного вектора на клетку.
После этого мальчику провели миелоабляцию (уничтожение кроветворных клеток) бусульфаном. Через два дня ему ввели модифицированные клетки.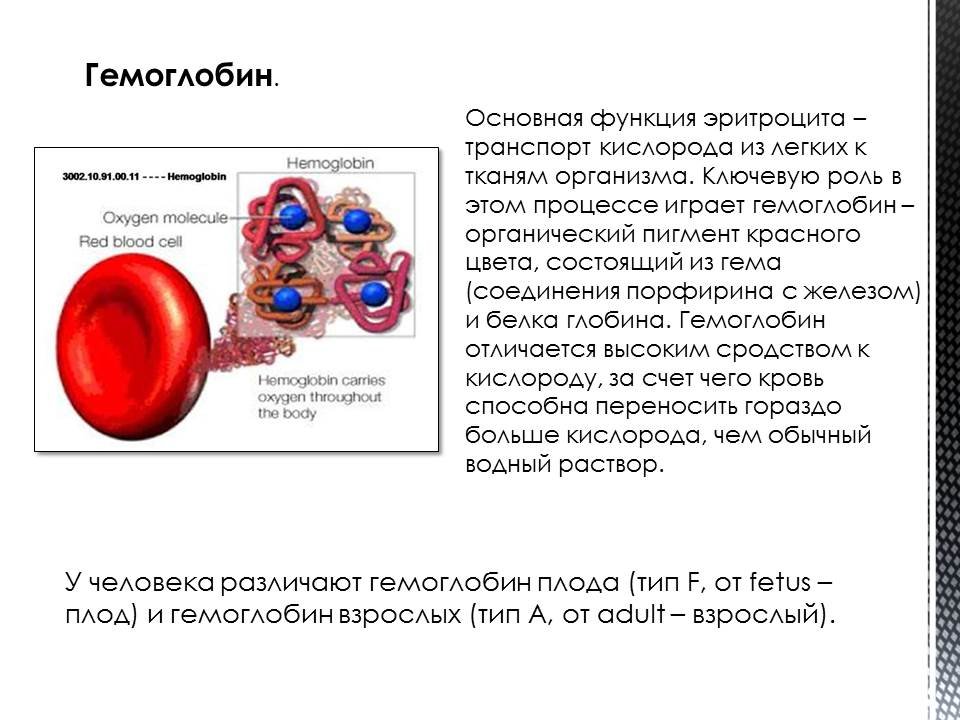 На всем протяжении лечения пациенту переливали кровь, пока уровень βA-T87Q не достиг от 25 до 30 процентов от всего гемоглобина на 88 день терапии.
На всем протяжении лечения пациенту переливали кровь, пока уровень βA-T87Q не достиг от 25 до 30 процентов от всего гемоглобина на 88 день терапии.
На шестой месяц уровень общего гемоглобина в крови мальчика стабилизировался на показателях от 106 до 120 граммов на литр (около нижней границы нормы). К девятому месяцу после введения модифицированных клеток βA-T87Q составлял 46 процентов, а к 15 месяцу — 48 процентов от всего гемоглобина.
В течении более чем 15 месяцев наблюдения у пациента не наблюдалось никаких клинических событий, связанных с серповидноклеточной анемией, показатели анализа крови нормализовались. По итогам наблюдения всю медикаментозную терапию отменили за ненадобностью. Основные побочные эффекты были связаны с миелоабляцией и устранены в процессе лечения.
Помимо описываемого пациента к настоящему времени терапию препаратом LentiGlobin BB305 получили еще несколько десятков французских и американских участников испытаний с серповидноклеточной анемией и другим наследственным дефектом бета-глобина — бета-талассемией.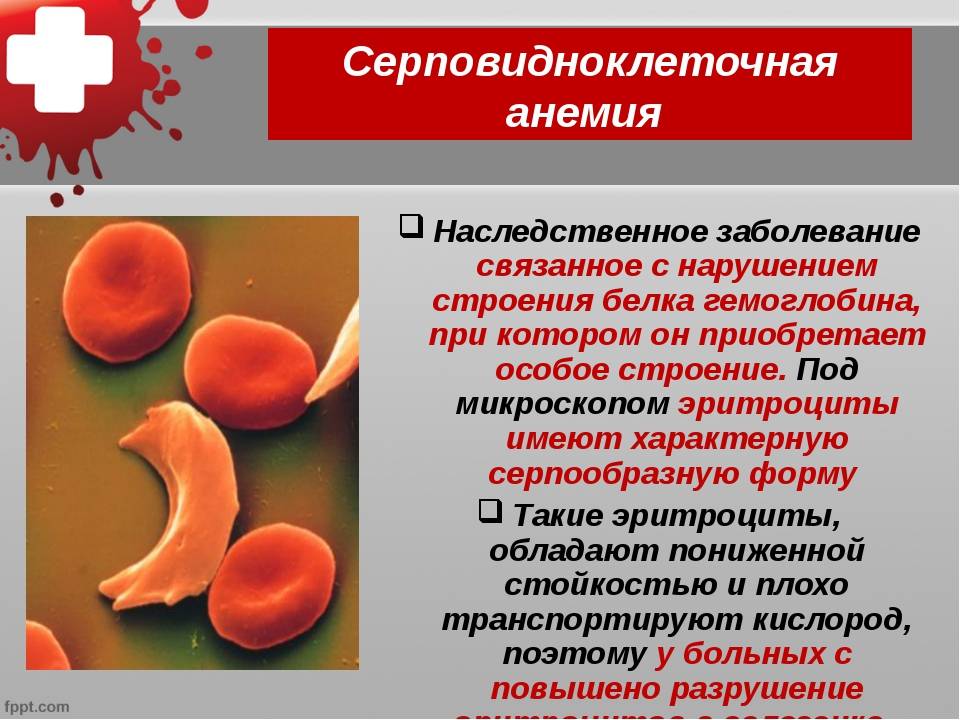 Предварительные результаты лечения признаны обнадеживающими, однако окончательные выводы делать пока рано, пишут авторы работы.
Предварительные результаты лечения признаны обнадеживающими, однако окончательные выводы делать пока рано, пишут авторы работы.
Недавно были разработаны несколько других экспериментальных подходов к терапии серповидноклеточной анемии и других гемоглобинопатий. В частности, ученые успешно испытали на мышах и клетках человека редактирование дефектного гена с помощью системы CRISPR/Cas9, убедились, что вещества, увеличивающие объем эритроцитов, препятствуют их деформации, и разработали метод генетической коррекции мутаций гемоглобина, основанный на пептидонуклеиновых кислотах.
Олег Лищук
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Серповидноклеточная анемия (SCD) представляет собой группу наследственных заболеваний эритроцитов. Красные кровяные тельца содержат гемоглобин, белок, переносящий кислород. Здоровые эритроциты имеют круглую форму и перемещаются по мелким кровеносным сосудам, доставляя кислород ко всем частям тела. У человека с ВСС гемоглобин ненормальный, из-за чего эритроциты становятся твердыми и липкими и похожи на С-образный сельскохозяйственный инструмент, называемый «серпом». Серповидные клетки рано погибают, что вызывает постоянную нехватку эритроцитов. Кроме того, когда они проходят через мелкие кровеносные сосуды, они застревают и блокируют кровоток. Это может вызвать боль и другие серьезные осложнения (проблемы со здоровьем), такие как инфекция, острый грудной синдром и инсульт.
У человека с ВСС гемоглобин ненормальный, из-за чего эритроциты становятся твердыми и липкими и похожи на С-образный сельскохозяйственный инструмент, называемый «серпом». Серповидные клетки рано погибают, что вызывает постоянную нехватку эритроцитов. Кроме того, когда они проходят через мелкие кровеносные сосуды, они застревают и блокируют кровоток. Это может вызвать боль и другие серьезные осложнения (проблемы со здоровьем), такие как инфекция, острый грудной синдром и инсульт.
Существует несколько типов SCD. Конкретный тип ВСС у человека зависит от генов, которые он унаследовал от своих родителей. Люди с ВСС наследуют гены, содержащие инструкции или код аномального гемоглобина.
Люди с этой формой ВСС наследуют два гена, по одному от каждого родителя, которые кодируют гемоглобин «S». Гемоглобин S представляет собой аномальную форму гемоглобина, из-за которой эритроциты становятся жесткими и серповидными.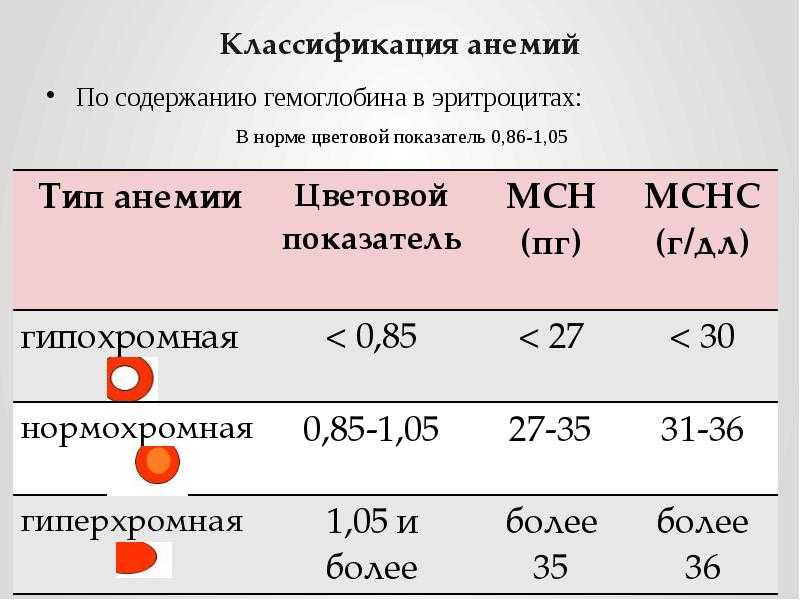 Это обычно называют серповидно-клеточная анемия и обычно является самой тяжелой формой заболевания.
Это обычно называют серповидно-клеточная анемия и обычно является самой тяжелой формой заболевания.
Люди с этой формой ВСС наследуют ген гемоглобина «S» от одного родителя и ген другого типа аномального гемоглобина, называемый «С», от другого родителя. Обычно это более легкая форма ВСС.
Инфографика: 5 фактов о серповидноклеточной анемии, которые следует знать
Знаете ли вы, что ВСС поражает людей во многих частях мира?
Люди с этой формой ВСС наследуют ген гемоглобина «S» от одного родителя и ген бета-талассемии, другого типа аномалии гемоглобина, от другого родителя. Существует два типа бета-талассемии: «нулевая» (HbS бета 0 ) и «плюсовая» (HbS бета + ). Те, у кого HbS бета 0 -талассемия, обычно имеют тяжелую форму ВСС. Люди с HbS бета + -талассемией, как правило, имеют более легкую форму ВСС.
Люди с этими формами ВСС наследуют один ген гемоглобина «S» и один ген, кодирующий другой аномальный тип гемоглобина («D», «E» или «O»). Тяжесть этих более редких типов ВСС варьируется.
Тяжесть этих более редких типов ВСС варьируется.
Люди с признаком серповидноклеточной анемии (SCT) наследуют ген гемоглобина «S» от одного родителя и нормальный ген (кодирующий гемоглобин «А») от другого другой родитель. Люди с SCT обычно не имеют каких-либо признаков заболевания. Однако в редких случаях у человека с ПКТ могут возникнуть проблемы со здоровьем; это чаще всего происходит, когда организм подвергается другим стрессам, например, когда человек обезвоживается или интенсивно тренируется. Кроме того, люди с SCT могут передать аномальный ген гемоглобина «S» своим детям.
Узнайте больше о признаке серповидно-клеточной анемии »
ВСС — это генетическое заболевание, присутствующее при рождении. Он передается по наследству, когда ребенок получает два гена — по одному от каждого родителя — которые кодируют аномальный гемоглобин.
ВСС диагностируется с помощью простого анализа крови. У детей, рожденных в США, чаще всего его обнаруживают при рождении во время рутинных скрининговых обследований новорожденных в больнице. Кроме того, ВСС может быть диагностирована еще в утробе матери. Диагностические тесты до рождения ребенка, такие как забор ворсин хориона и амниоцентез, могут выявить хромосомные или генетические аномалии у ребенка. При взятии проб ворсин хориона исследуется крошечный кусочек плаценты, называемый ворсинами хориона. Амниоцентез исследует небольшой образец амниотической жидкости, окружающей ребенка.
У детей, рожденных в США, чаще всего его обнаруживают при рождении во время рутинных скрининговых обследований новорожденных в больнице. Кроме того, ВСС может быть диагностирована еще в утробе матери. Диагностические тесты до рождения ребенка, такие как забор ворсин хориона и амниоцентез, могут выявить хромосомные или генетические аномалии у ребенка. При взятии проб ворсин хориона исследуется крошечный кусочек плаценты, называемый ворсинами хориона. Амниоцентез исследует небольшой образец амниотической жидкости, окружающей ребенка.
Поскольку дети с ВСС подвержены повышенному риску инфицирования и других проблем со здоровьем, важное значение имеют ранняя диагностика и лечение.
Поговорите со своим врачом, чтобы узнать, как сдать анализ, и объяснить результаты после тестирования.
SCD Fact Sheet
Посмотреть и распечатать
У людей с SCD могут появиться признаки болезни в течение первого года жизни, обычно в возрасте около 5 месяцев. Симптомы и осложнения ВСС различны для каждого человека и могут варьироваться от легких до тяжелых. Узнайте об осложнениях.
Симптомы и осложнения ВСС различны для каждого человека и могут варьироваться от легких до тяжелых. Узнайте об осложнениях.
Общие стратегии профилактики
Лечение ВСС направлено на профилактику и лечение эпизодов боли и других осложнений. Стратегии профилактики включают изменение образа жизни, а также медицинский осмотр и вмешательства для предотвращения осложнений ВСС.
Образ жизни
Существуют простые шаги, которые люди с ВСС могут предпринять, чтобы предотвратить и уменьшить возникновение болевых кризисов, в том числе следующие:

Простые шаги по предотвращению вредоносных инфекций включают следующее:
Медицинские обследования и вмешательства для предотвращения осложнений ВСС
Профилактика инфекций
 Пенициллин для ежедневного приема обычно не назначают детям с другими типами ВСС, за исключением случаев, когда тяжесть заболевания аналогична тяжести HbSS, например HbS бета 9.0027 0 -талассемия.
Пенициллин для ежедневного приема обычно не назначают детям с другими типами ВСС, за исключением случаев, когда тяжесть заболевания аналогична тяжести HbSS, например HbS бета 9.0027 0 -талассемия. Профилактика потери зрения
Профилактика инсульта
Профилактика тяжелой анемии

Лечение болевых кризисов
При возникновении болевых кризов клиническое лечение может включать следующее:
Специальные методы лечения для предотвращения осложнений ВСС
ВСС — это заболевание, которое со временем ухудшается. Доступны методы лечения, которые могут предотвратить осложнения и продлить жизнь тем, у кого есть это заболевание. Эти варианты лечения и их эффекты могут быть разными для каждого человека, в зависимости от симптомов и тяжести заболевания. Важно понимать преимущества и риски каждого варианта лечения. В настоящее время FDA одобрило четыре метода лечения ВСС 9.0027 [1] .

Недавно были разработаны несколько других методов лечения ВСС, которые все еще проходят клинические испытания и, следовательно, еще не одобрены FDA.
Единственная одобренная FDA терапия, которая может быть в состоянии вылечить ВСС, — это трансплантация костного мозга или стволовых клеток.
Костный мозг представляет собой мягкую жировую ткань в центре костей, где образуются клетки крови.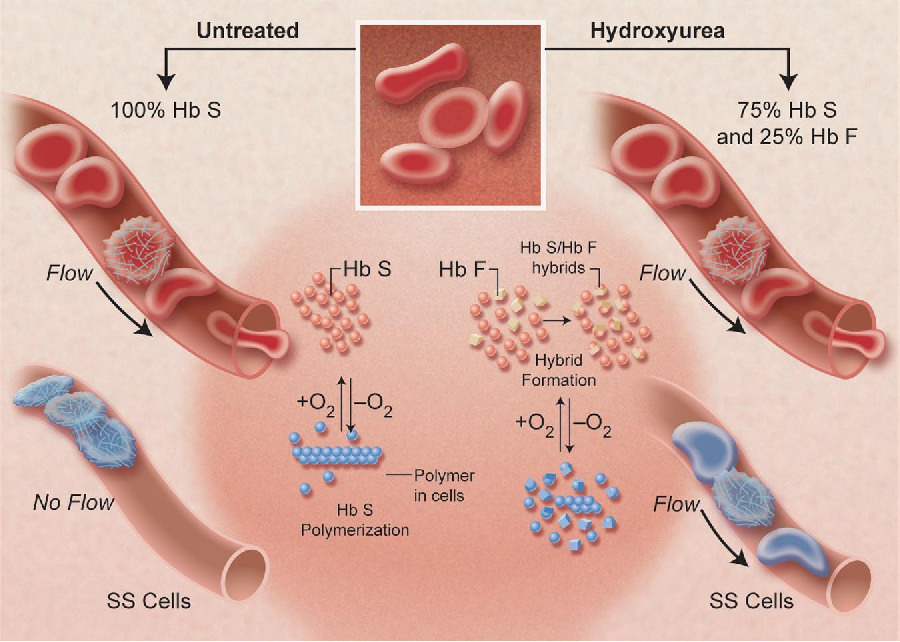 Трансплантация костного мозга или стволовых клеток — это процедура, при которой здоровые клетки, образующие кровь, берутся у одного человека — донора — и пересаживаются человеку, чей костный мозг не работает должным образом.
Трансплантация костного мозга или стволовых клеток — это процедура, при которой здоровые клетки, образующие кровь, берутся у одного человека — донора — и пересаживаются человеку, чей костный мозг не работает должным образом.
Трансплантация костного мозга или стволовых клеток очень опасна и может иметь серьезные побочные эффекты, включая смерть. Чтобы трансплантат сработал, костный мозг должен быть близким. Обычно лучшим донором является брат или сестра. Трансплантация костного мозга или стволовых клеток наиболее распространена в случаях тяжелой ВСС у детей с минимальным поражением органов в результате заболевания.
[1] CDC будет периодически пересматривать и обновлять этот список лечения, когда новые методы лечения будут одобрены FDA.
Серповидноклеточная анемия — это группа заболеваний, поражающих гемоглобин, молекулу эритроцитов, доставляющую кислород к клеткам по всему телу. Люди с этим заболеванием имеют атипичные молекулы гемоглобина, называемые гемоглобином S, которые могут деформировать эритроциты в серповидную или серповидную форму.
Признаки и симптомы серповидно-клеточной анемии обычно проявляются в раннем детстве. Характерные черты этого расстройства включают низкое количество эритроцитов (анемия), повторные инфекции и периодические приступы боли. Тяжесть симптомов варьируется от человека к человеку. Некоторые люди имеют легкие симптомы, в то время как других часто госпитализируют с более серьезными осложнениями.
Признаки и симптомы серповидноклеточной анемии вызваны серповидностью эритроцитов. Когда эритроциты желтеют, они преждевременно разрушаются, что может привести к анемии. Анемия может вызвать одышку, утомляемость и задержку роста и развития у детей.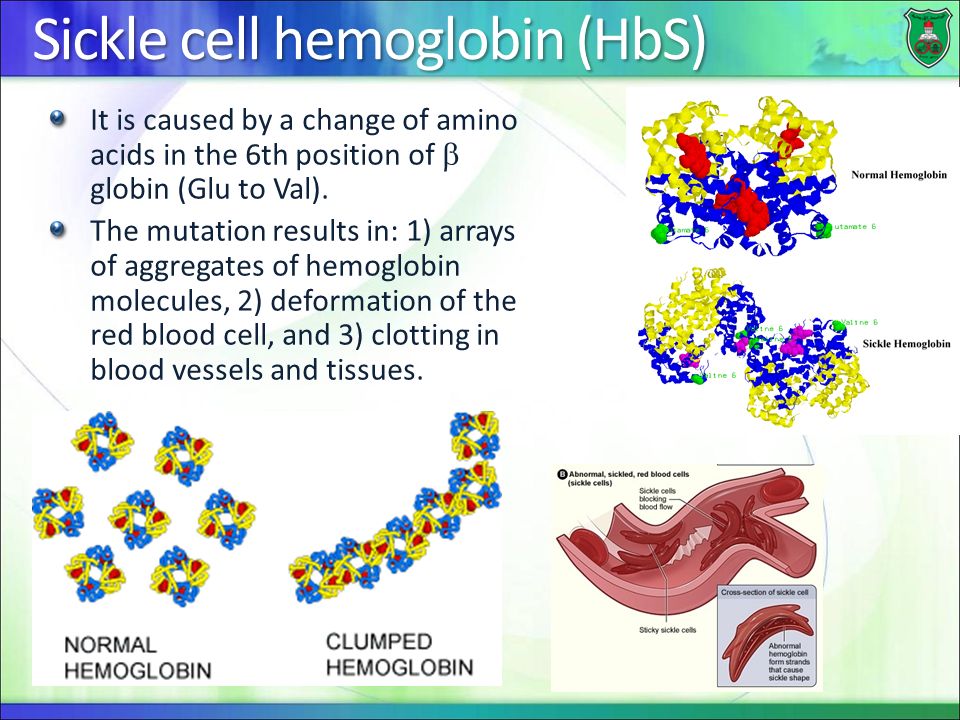 Быстрое разрушение эритроцитов также может вызвать пожелтение глаз и кожи, что является признаком желтухи. Болезненные эпизоды могут возникать, когда серповидные эритроциты, жесткие и негибкие, застревают в мелких кровеносных сосудах. Эти эпизоды лишают ткани и органы, такие как легкие, почки, селезенка и мозг, богатой кислородом крови и могут привести к повреждению органов. Особенно серьезным осложнением серповидно-клеточной анемии является высокое кровяное давление в кровеносных сосудах, снабжающих легкие (легочная гипертензия), что может привести к сердечной недостаточности. Легочная гипертензия встречается примерно у 10 процентов взрослых с серповидно-клеточной анемией.
Быстрое разрушение эритроцитов также может вызвать пожелтение глаз и кожи, что является признаком желтухи. Болезненные эпизоды могут возникать, когда серповидные эритроциты, жесткие и негибкие, застревают в мелких кровеносных сосудах. Эти эпизоды лишают ткани и органы, такие как легкие, почки, селезенка и мозг, богатой кислородом крови и могут привести к повреждению органов. Особенно серьезным осложнением серповидно-клеточной анемии является высокое кровяное давление в кровеносных сосудах, снабжающих легкие (легочная гипертензия), что может привести к сердечной недостаточности. Легочная гипертензия встречается примерно у 10 процентов взрослых с серповидно-клеточной анемией.
Серповидно-клеточная анемия поражает миллионы людей во всем мире. Это наиболее распространено среди людей, предки которых происходят из Африки; Средиземноморские страны, такие как Греция, Турция и Италия; Аравийский полуостров; Индия; и испаноязычные регионы Южной Америки, Центральной Америки и некоторых частей Карибского бассейна.
Серповидно-клеточная анемия является наиболее распространенным наследственным заболеванием крови в Соединенных Штатах, которым страдают около 100 000 американцев. По оценкам, это заболевание встречается у 1 из 500 афроамериканцев и у 1 из 1000–1400 латиноамериканцев.
Мутации в гене HBB вызывают серповидноклеточную анемию. Ген HBB обеспечивает инструкции по созданию одной части гемоглобина. Гемоглобин состоит из четырех белковых субъединиц, обычно двух субъединиц, называемых альфа-глобином, и двух субъединиц, называемых бета-глобином. Ген HBB предоставляет инструкции по производству бета-глобина. Различные версии бета-глобина являются результатом различных мутаций в гене HBB . Один конкретный HBB 9Мутация гена 0230 приводит к образованию аномальной версии бета-глобина, известной как гемоглобин S (HbS). Другие мутации в гене HBB приводят к дополнительным аномальным версиям бета-глобина, таким как гемоглобин C (HbC) и гемоглобин E (HbE).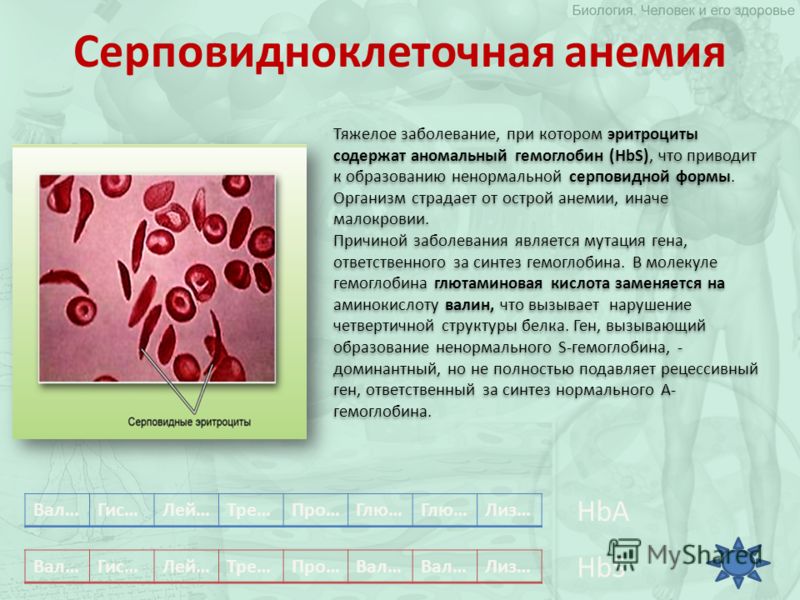 Мутации гена HBB также могут приводить к необычно низкому уровню бета-глобина; эта аномалия называется бета-талассемией.
Мутации гена HBB также могут приводить к необычно низкому уровню бета-глобина; эта аномалия называется бета-талассемией.
У людей с серповидноклеточной анемией по крайней мере одна из субъединиц бета-глобина в гемоглобине заменена гемоглобином S. При серповидноклеточной анемии (также называемой гомозиготной серповидноклеточной анемией), которая является наиболее распространенной формой серповидноклеточной анемии, гемоглобин S заменяет обе субъединицы бета-глобина в гемоглобине. При других типах серповидноклеточной анемии только одна субъединица бета-глобина в гемоглобине заменяется гемоглобином S. Другая субъединица бета-глобина заменяется другим аномальным вариантом, таким как гемоглобин C. Например, люди с серповидноклеточным гемоглобином C (HbSC) имеют молекулы гемоглобина с гемоглобином S и гемоглобином C вместо бета-глобина. Если мутации, которые продуцируют гемоглобин S и бета-талассемию, происходят вместе, у людей есть гемоглобин S-бета-талассемия (HbSBetathal).
Аномальные версии бета-глобина могут деформировать эритроциты в серповидную форму. Серповидные эритроциты преждевременно погибают, что может привести к анемии. Иногда негибкие серповидные клетки застревают в мелких кровеносных сосудах и могут вызвать серьезные медицинские осложнения.
Это состояние наследуется по аутосомно-рецессивному типу, что означает, что обе копии гена в каждой клетке имеют мутации. Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания.
 gov
gov