


2011-2017 © МБУЗ ГКП № 7, г.Челябинск.
Врожденные мышечные дистрофии (ВМД) — группа генетически гетерогенных заболеваний, характеризующихся диффузной мышечной гипотонией, гипорефлексией, снижением объема активных движений, возникающих с рождения или в период новорожденности. К настоящему времени идентифицировано более десяти генов, ответственных за возникновение изолированных форм ВМД, и охарактеризованы белковые продукты их экспрессии [1]. Один из самых редких вариантов этой группы заболеваний обусловлен мутациями в гене LMNA, картированном на хромосоме 1q11—q23 [2] и состоящем из 12 экзонов [3]. Кодируемый им белок преламин, А является предшественником зрелых форм ламинов, А и С, которые формируют структуру внутренней мембраны ядра клетки.
Описано одиннадцать основных фенотипов, обусловленных мутациями в гене LMNA [4—7]. Наиболее часто они приводят к формированию прогрессирующих мышечных дистрофий (ПМД) с аутосомно-доминантным типом наследования, которые имеют различный возраст манифестации и особенности клинических проявлений: ПМД Эмери—Дрейфуса 2-го типа (OMIM:181350), поясно-конечностная ПМД 1 В типа (OMIM:159001) и ВМД (OMIM:613205). В ряде работ были сделаны попытки объяснения особенностей течения ПМД на основании различий в характере мутаций в гене LMNA. Так, некоторые авторы показали, что к возникновению более тяжелых фенотипов приводят мутации, обладающие доминантно-негативным эффектом, в то время как у больных с наличием миссенс-мутаций в этом гене, приводящих только к гаплонедостаточности, отмечается более мягкое течение заболевания [8]. Однако E. Mercuri и соавт. [9] описали 5 больных с различными вариантами ПМД, возникающими с рождения до взрослого возраста, у которых была обнаружена одна и та же миссенс-мутация Gln358Lys.
Первые сведения о единичных случаях этой формы ПМД были опубликованы в 2004 г. E. Mercuri и соавт. [9] и 2005 г. A. D`Amico и соавт. [10]. Однако выделение врожденной ламинопатии в отдельный вариант ВМД было предложено в 2008 г. S. Quijano-Roy и соавт. [11], которые суммировали клинико-генетические характеристики 15 больных с ВМД, обусловленными мутациями в гене LMNA.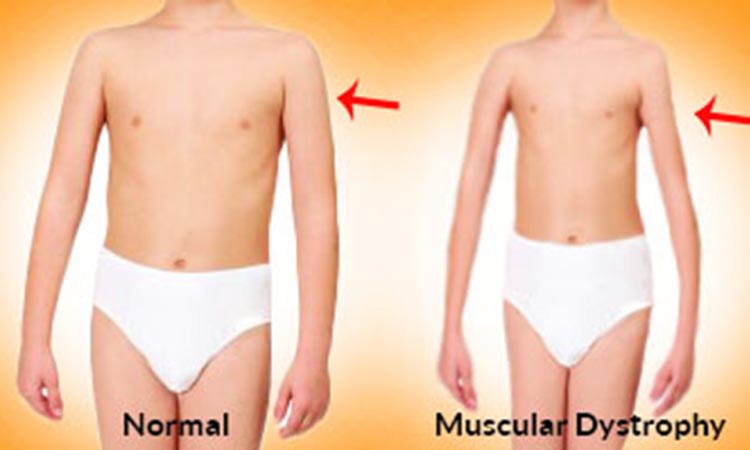 К настоящему времени описано более 40 больных с этим вариантом ВМД из популяций Италии, Франции, Бразилии, Германии и др. [6, 11—15].
К настоящему времени описано более 40 больных с этим вариантом ВМД из популяций Италии, Франции, Бразилии, Германии и др. [6, 11—15].
Цель настоящей работы — представление клинико-генетических характеристики 5 больных с ВМД, обусловленной мутациями в гене LMNA, впервые выявленных в России.
Были исследованы образцы ДНК 42 пробандов, наблюдающихся в ФБГНУ «Медико-генетический научный центр», с характерными признаками ВМД в возрасте от 2 мес до 9 лет из неродственных семей, проживающих на территории Р.Ф. Диагноз устанавливали на основании данных генеалогического анализа, неврологического осмотра, показателей активности креатинфосфокиназы (КФК) в сыворотке крови, данных электронейромиографии (ЭНМГ). Большинство больных имели результаты проведенной ранее магнитно-резонансной томографии (МРТ) головного мозга и исследования мышечного биоптата.
Выделение геномной ДНК проводили из лимфоцитов периферической крови с помощью набора реактивов DLAtomTM DNA Prep100 («Isogene Lab. ltd.», Россия) по протоколу производителя. Исследуемые участки ДНК амплифицировали методом ПЦР с использованием уникальных праймеров, дизайн которых выполнен в лаборатории ДНК-диагностики, синтез — в ЗАО «Евроген». Качественную визуализацию продуктов ПЦР-реакции осуществляли при помощи электрофореза в 7% полиакриламидном геле (ПААГ) с соотношением акриамида и бисакриламида 29:1. Определение нуклеотидной последовательности всей кодирующей области и участков экзон-интронных соединений гена LMNA проводили при помощи метода прямого автоматического секвенирования на приборе ABI Prism 3100 («Applied Biosystems», США) с использованием протокола фирмы-производителя. Полученные данные анализировали при помощи программы Chromas 2.4.1.
ltd.», Россия) по протоколу производителя. Исследуемые участки ДНК амплифицировали методом ПЦР с использованием уникальных праймеров, дизайн которых выполнен в лаборатории ДНК-диагностики, синтез — в ЗАО «Евроген». Качественную визуализацию продуктов ПЦР-реакции осуществляли при помощи электрофореза в 7% полиакриламидном геле (ПААГ) с соотношением акриамида и бисакриламида 29:1. Определение нуклеотидной последовательности всей кодирующей области и участков экзон-интронных соединений гена LMNA проводили при помощи метода прямого автоматического секвенирования на приборе ABI Prism 3100 («Applied Biosystems», США) с использованием протокола фирмы-производителя. Полученные данные анализировали при помощи программы Chromas 2.4.1.
В результате проведения ДНК-диагностики, направленной на поиск мутаций в гене LMNA, выявлено 5 больных (4 мальчика и 1 девочка) в возрасте от 4 мес до 7 лет. Клинико-генетические характеристики больных с ВМД, обусловленной мутациями в гене
LMNA, представлены в таблице.
Клинико-генетические характеристики больных ВМД, обусловленной мутациями в гене LMNA
Как видно из таблицы, все больные имели классические проявления врожденной ламинопатии. Однако у 1 из них имелись отличия в возрасте манифестации, тяжести течения заболевания и типе мутации, что позволило разделить наблюдаемых нами больных на 2 группы. Это коррелирует с данными других исследователей, поскольку, по мнению ряда авторов [5, 7, 11, 13], существуют два клинических варианта ВМД, обусловленных мутациями в гене LMNA, отличающихся по возрасту начала и тяжести течения заболевания.
Первую группу составили 4 больных (3 мальчика и 1 девочка) с тяжелой формой болезни имели отягощенный акушерский анамнез и страдали с рождения. На момент осмотра у всех отмечались выраженная гипотрофия, гипотония, мышечная слабость с преимущественным поражением аксиальных мышц и мышц лопаточно-плечевого региона, что приводило к возникновению симптома «свисающей головы». В основном их моторное развитие было минимальным. Лишь 1 мальчик мог передвигаться с поддержкой с 18 мес до 4 лет. Достаточно рано у всех больных этой группы появлялась тугоподвижность в голеностопных суставах, что в последующем приводило к возникновению фиксированной сгибательной контрактуры, а по мере течения заболевания контрактуры формировались и в других крупных суставах (в том числе в локтевых — у 3 больных). Нарушение сердечного ритма имели 2 больных. Дыхательные нарушения, требующие специальной поддержки, наблюдались только у 5-летней больной, которой установили трахеостому в 4,5 года. Однако некоторые авторы [13] считают, что возникновение дыхательной недостаточности с необходимостью использования вспомогательных средств характерно для всех больных с этим вариантом ВМД. У 2 больных на МРТ головного мозга были выявлены признаки перивентрикулярной лейкоэнцефалопатии. Хотя в литературе есть описание [16] 1 больного с ВМД, обусловленной мутацией в гене
LMNA, также имевшего локально повышенный, неспецифический МР-сигнал от белого вещества мозга, мы склонны считать, что в нашем случае выявленные признаки перивентрикулярной лейкоэнцефалопатии, вероятнее всего, были обусловлены присоединившимся перинатальным гипоксически-имшемическим поражением ЦНС.
В основном их моторное развитие было минимальным. Лишь 1 мальчик мог передвигаться с поддержкой с 18 мес до 4 лет. Достаточно рано у всех больных этой группы появлялась тугоподвижность в голеностопных суставах, что в последующем приводило к возникновению фиксированной сгибательной контрактуры, а по мере течения заболевания контрактуры формировались и в других крупных суставах (в том числе в локтевых — у 3 больных). Нарушение сердечного ритма имели 2 больных. Дыхательные нарушения, требующие специальной поддержки, наблюдались только у 5-летней больной, которой установили трахеостому в 4,5 года. Однако некоторые авторы [13] считают, что возникновение дыхательной недостаточности с необходимостью использования вспомогательных средств характерно для всех больных с этим вариантом ВМД. У 2 больных на МРТ головного мозга были выявлены признаки перивентрикулярной лейкоэнцефалопатии. Хотя в литературе есть описание [16] 1 больного с ВМД, обусловленной мутацией в гене
LMNA, также имевшего локально повышенный, неспецифический МР-сигнал от белого вещества мозга, мы склонны считать, что в нашем случае выявленные признаки перивентрикулярной лейкоэнцефалопатии, вероятнее всего, были обусловлены присоединившимся перинатальным гипоксически-имшемическим поражением ЦНС.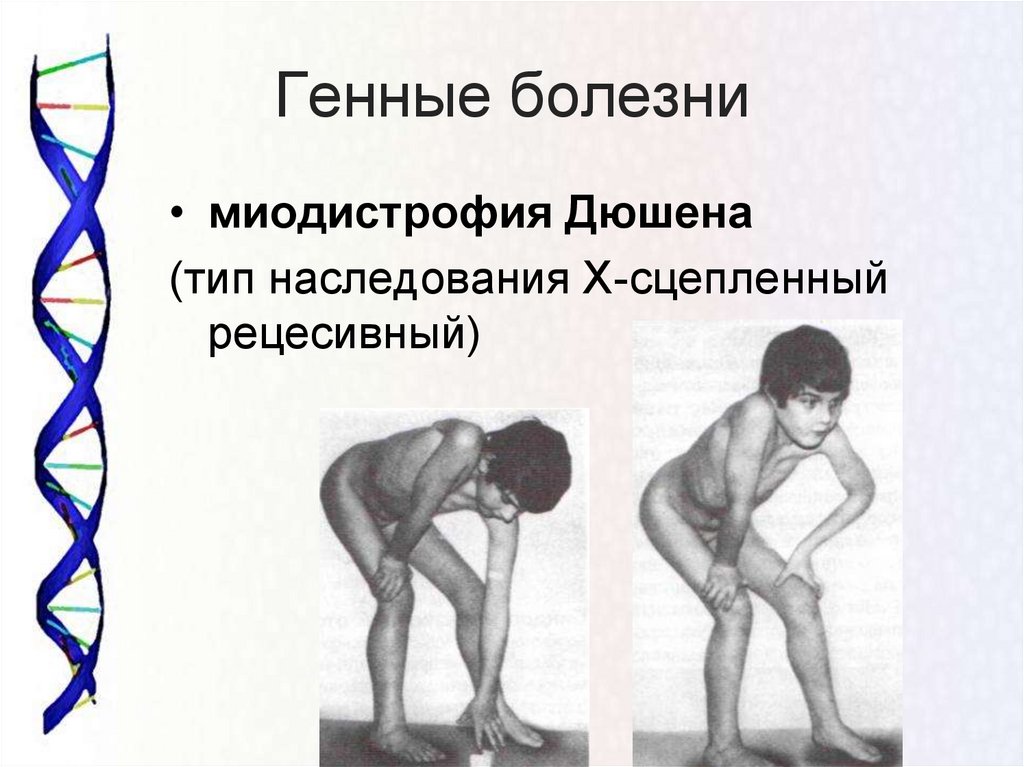 У одного пробанда в ранее проведенном исследовании мышечного биоптата были выявлены признаки митохондриальной миопатии, а у другого — неспецифического воспалительного процесса в исследуемой мышце. В литературе [11, 17, 18] также имеются сведения о сходном характере изменений в мышечных биоптатах у некоторых больных с этой формой ВМД. 2 мальчика из этой группы внезапно скончались в возрасте 10 мес и 9 лет. У всех больных в этой группе при проведении ДНК-диагностики была обнаружена мутация c.94_96delAAG в гетерозиготном состоянии в 1-м экзоне гена LMNA, приводящая к делеции аминокислоты лизин (Lys) в 32-м положении полипептидной цепи. Впервые эту мутацию обнаружили M. Vytopil и соавт. [19] в 2002 г. у больной с тяжелой клинической картиной ПМД Эмери—Дрейфуса. Авторы показали, что женщина унаследовала ее от отца, который в возрасте 52 лет был здоров (на основании чего авторы предположили наличие неполной пенетрантности гена при наличии данной мутации). В литературе также имеются описания больных с другими фенотипами ПМД, манифестирующих с периода новорожденности [10] или в юношеском и взрослом возрасте [20, 21], обусловленными мутацией c.
У одного пробанда в ранее проведенном исследовании мышечного биоптата были выявлены признаки митохондриальной миопатии, а у другого — неспецифического воспалительного процесса в исследуемой мышце. В литературе [11, 17, 18] также имеются сведения о сходном характере изменений в мышечных биоптатах у некоторых больных с этой формой ВМД. 2 мальчика из этой группы внезапно скончались в возрасте 10 мес и 9 лет. У всех больных в этой группе при проведении ДНК-диагностики была обнаружена мутация c.94_96delAAG в гетерозиготном состоянии в 1-м экзоне гена LMNA, приводящая к делеции аминокислоты лизин (Lys) в 32-м положении полипептидной цепи. Впервые эту мутацию обнаружили M. Vytopil и соавт. [19] в 2002 г. у больной с тяжелой клинической картиной ПМД Эмери—Дрейфуса. Авторы показали, что женщина унаследовала ее от отца, который в возрасте 52 лет был здоров (на основании чего авторы предположили наличие неполной пенетрантности гена при наличии данной мутации). В литературе также имеются описания больных с другими фенотипами ПМД, манифестирующих с периода новорожденности [10] или в юношеском и взрослом возрасте [20, 21], обусловленными мутацией c.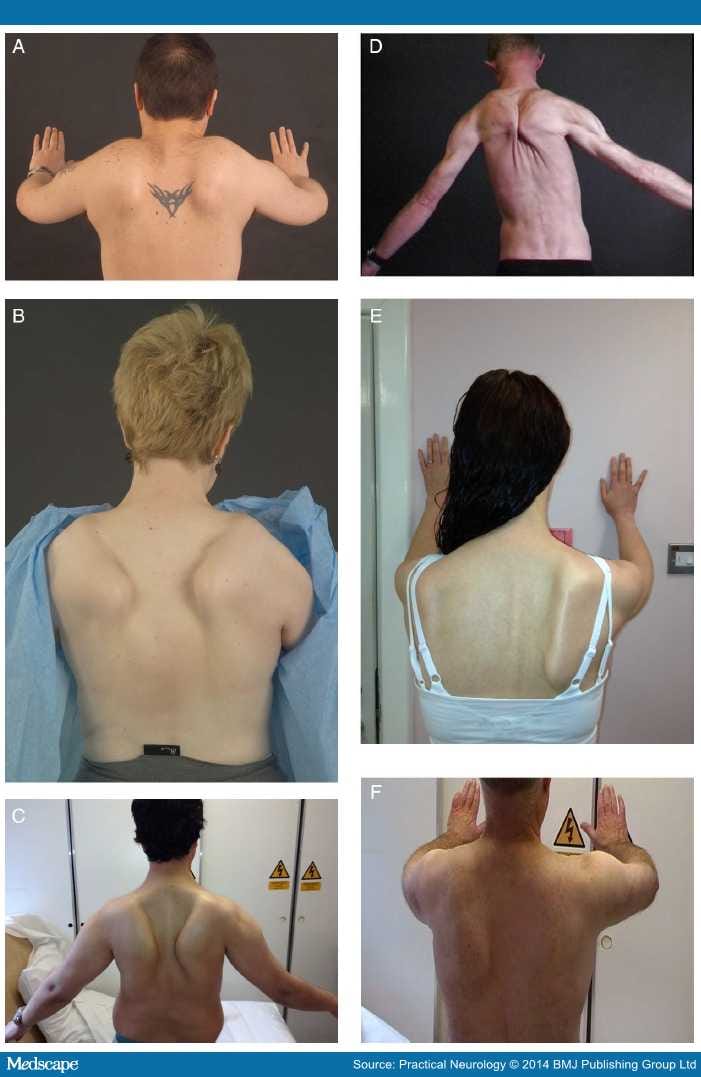 94_96delAAG в гене LMNA.
94_96delAAG в гене LMNA.
Представляем описание 2 клинических случаев из этой группы больных ВМД.
Девочка Б-на, впервые осмотрена в 13 мес по поводу жалоб на выраженную мышечную гипотонию с рождения и задержку темпов моторного развития. Из анамнеза известно, что ребенок от первой беременности, протекавшей в I триместре на фоне токсикоза и фетоплацентарной недостаточности, с формированием внутриутробной гипотрофии плода в III триместре. Роды срочные, самостоятельные на 39-й неделе, с использованием медикаментозной стимуляции и приемов выдавливания плода. При рождении — вес 2800 г, длина 49 см, по шкале Апгар 5/7 баллов за счет аспирации мекониальными водами. Получала ИВЛ в течение 5 сут. Домой выписана на 22-е сутки. С рождения отмечался симптомокомплекс вялого ребенка. На МРТ головного мозга, проведенной в периоде новорожденности, выявлены диффузная субкортикальная субатрофия головного мозга и перивентрикулярная лейкоэнцефалопатия. На ЭНМГ: первично-мышечный уровень поражения. При объективном осмотре в 13 мес: выраженная диффузная мышечная гипотрофия, гипотония с минимальной двигательной активностью в верхних и нижних конечностях, сухожильная арефлексия. Выраженная слабость аксиальных мышц. Симптом «свисающей головы». Контрактур нет. Психоэмоциональная сфера по возрасту. Уровень КФК в сыворотке крови 2000 ед/л. В 15 мес при исследовании мышечного биоптата выявлены признаки неспецифического воспалительного процесса, по поводу чего была предпринята безуспешная попытка терапии гормональными препаратами. При осмотре в 20 мес — голова свисает, удерживать ее может только при подъеме и фиксации плеч, не может поднять руки выше горизонтального уровня и подтянуть ноги к животу, сухожильные рефлексы не вызываются. Контрактур нет. Говорит несколько слов. Уровень КФК в сыворотке крови снизился до 700 ед/л. В возрасте 29 мес родители отметили появление контрактур в голеностопных суставах. При объективном осмотре в 3 года 8 мес — не садится, не стоит, не ходит, сидит с опорой, лицо «миопата», выраженная диффузная гипотрофия и гипотония, голову не удерживает, симптом «свисающей головы», повороты головы и поднятие плеч ограничены, «крыловидные лопатки», выраженный гиперлордоз и сколиоз поясничного отдела позвоночника.
При объективном осмотре в 13 мес: выраженная диффузная мышечная гипотрофия, гипотония с минимальной двигательной активностью в верхних и нижних конечностях, сухожильная арефлексия. Выраженная слабость аксиальных мышц. Симптом «свисающей головы». Контрактур нет. Психоэмоциональная сфера по возрасту. Уровень КФК в сыворотке крови 2000 ед/л. В 15 мес при исследовании мышечного биоптата выявлены признаки неспецифического воспалительного процесса, по поводу чего была предпринята безуспешная попытка терапии гормональными препаратами. При осмотре в 20 мес — голова свисает, удерживать ее может только при подъеме и фиксации плеч, не может поднять руки выше горизонтального уровня и подтянуть ноги к животу, сухожильные рефлексы не вызываются. Контрактур нет. Говорит несколько слов. Уровень КФК в сыворотке крови снизился до 700 ед/л. В возрасте 29 мес родители отметили появление контрактур в голеностопных суставах. При объективном осмотре в 3 года 8 мес — не садится, не стоит, не ходит, сидит с опорой, лицо «миопата», выраженная диффузная гипотрофия и гипотония, голову не удерживает, симптом «свисающей головы», повороты головы и поднятие плеч ограничены, «крыловидные лопатки», выраженный гиперлордоз и сколиоз поясничного отдела позвоночника. Контрактуры в голеностопных, коленных, тазобедренных, локтевых, межфаланговых суставах. Уровень КФК сыворотки крови составляет 441 ед/л. На ЭКГ: незначительная синусовая тахикардия, аритмия с частотой сердечных сокращений 103—130 ударов в 1 мин. Электрическая ось сердца отклонена вправо. Неполная блокада правой ножки пучка Гиса. При проведении ЭХО-кардиографии пороков сердца не выявлено. Полости сердца не расширены. Клапаны интактны, сократительная способность удовлетворительная. В 4,5 года появились дыхательные нарушения, требующие специальной поддержки — установлена трахеостома. При осмотре в 5 лет у девочки на фоне выраженной диффузной гипотрофии, гипотонии, сухожильной арефлексии имеет место ригидный поясничный лордосколиоз, сохраняющийся даже в положении лежа на спине, контрактуры во всех крупных и межфаланговых суставах. Трудности при глотании и жевании твердой пищи. Интеллект соответствует возрасту.
Контрактуры в голеностопных, коленных, тазобедренных, локтевых, межфаланговых суставах. Уровень КФК сыворотки крови составляет 441 ед/л. На ЭКГ: незначительная синусовая тахикардия, аритмия с частотой сердечных сокращений 103—130 ударов в 1 мин. Электрическая ось сердца отклонена вправо. Неполная блокада правой ножки пучка Гиса. При проведении ЭХО-кардиографии пороков сердца не выявлено. Полости сердца не расширены. Клапаны интактны, сократительная способность удовлетворительная. В 4,5 года появились дыхательные нарушения, требующие специальной поддержки — установлена трахеостома. При осмотре в 5 лет у девочки на фоне выраженной диффузной гипотрофии, гипотонии, сухожильной арефлексии имеет место ригидный поясничный лордосколиоз, сохраняющийся даже в положении лежа на спине, контрактуры во всех крупных и межфаланговых суставах. Трудности при глотании и жевании твердой пищи. Интеллект соответствует возрасту.
Мальчик К-в, осмотрен впервые в 7 лет. Из анамнеза известно, что ребенок от четвертой беременности, протекавшей с токсикозом. Роды вторые, срочные, самостоятельные. Вес при рождении 3200 г, длина 52 см, по шкале Апгар 7/8 баллов, закричал не сразу. На 3-и сутки установлен диагноз «врожденный порок сердца: дефект межжелудочковой перегородки», по поводу чего перенес успешное оперативное лечение в возрасте 5 мес. С рождения отмечался симтомокомплекс вялого ребенка. Голову держит с 8 мес, сам никогда не садился, ходил с поддержкой с 18 мес до 4 лет. С 6 мес развился эпилептический синдром в виде левосторонних гемиклоний и кивков, купированных по настоящее время приемом противоэпилептических препаратов (конвулекс до 30 мг/кг). При проведении биопсии мышц выявлены признаки митохондриальной миопатии. Уровень КФК в сыворотке крови больного варьировал от 2300 ед/л в начале заболевания до 489 ед/л в возрасте 5 лет. Объективно: диффузная гипотрофия, гипотония, больше выраженная в аксиальных мышцах, симптом «свисающей головы», «лицо миопата», сухожильная арефлексия, выраженный гиперлордоз в поясничном отделе позвоночника, сохраняющийся даже в положении лежа на спине, фиксированный сколиоз грудного отдела позвоночника.
Роды вторые, срочные, самостоятельные. Вес при рождении 3200 г, длина 52 см, по шкале Апгар 7/8 баллов, закричал не сразу. На 3-и сутки установлен диагноз «врожденный порок сердца: дефект межжелудочковой перегородки», по поводу чего перенес успешное оперативное лечение в возрасте 5 мес. С рождения отмечался симтомокомплекс вялого ребенка. Голову держит с 8 мес, сам никогда не садился, ходил с поддержкой с 18 мес до 4 лет. С 6 мес развился эпилептический синдром в виде левосторонних гемиклоний и кивков, купированных по настоящее время приемом противоэпилептических препаратов (конвулекс до 30 мг/кг). При проведении биопсии мышц выявлены признаки митохондриальной миопатии. Уровень КФК в сыворотке крови больного варьировал от 2300 ед/л в начале заболевания до 489 ед/л в возрасте 5 лет. Объективно: диффузная гипотрофия, гипотония, больше выраженная в аксиальных мышцах, симптом «свисающей головы», «лицо миопата», сухожильная арефлексия, выраженный гиперлордоз в поясничном отделе позвоночника, сохраняющийся даже в положении лежа на спине, фиксированный сколиоз грудного отдела позвоночника. Фиксированные контрактуры в голеностопных, коленных, тазобедренных суставах (рис. 1). Трудности при глотании и жевании твердой пищи, носовой оттенок голоса, псевдобульбарный парез. Интеллект соответствует возрасту.
Фиксированные контрактуры в голеностопных, коленных, тазобедренных суставах (рис. 1). Трудности при глотании и жевании твердой пищи, носовой оттенок голоса, псевдобульбарный парез. Интеллект соответствует возрасту.
Рис. 1. Больной К-в, 7 лет. Вид лежа на спине (а) и сидя (б).
Клинические проявления второго варианта BMD, обусловленной мутациями в гене LMNA (более легкая форма) по данным некоторых авторов [5, 7, 11, 13] появляются на 1-м году жизни после периода нормального моторного развития и, хотя также характеризуются прогрессирующей слабостью мышц аксиальных и поясов конечностей, больные более длительно могут передвигаться самостоятельно.
В одном случае и мы наблюдали такой вариант развития ВМД, обусловленный мутацией в гене LMNA. Мальчик Б-н, осмотрен в 5 лет по поводу жалоб на нарушение походки, общую слабость, трудности подъема с пола. Из анамнеза известно, что ребенок от девятой беременности, протекавшей на фоне угрозы прерывания. Роды третьи, самостоятельные, в срок. При рождении вес 4000 г, длина 53 см, закричал сразу. Домой выписан на 5-е сутки. Голову держит с 1,5 мес, сел самостоятельно в 15 мес, пошел в 18 мес. Первые признаки заболевания появились в 3 мес, когда родители обратили внимание на то, что ребенок стал хуже удерживать голову. При проведении ЭНМГ выявлен первично мышечный уровень поражения. На ЭКГ в 4 года выявлена выраженная синусовая тахикардия, частота сердечных сокращений до 112 ударов в 1 мин. Снижен вольтаж QRS в стандартных и усиленных отведениях. При проведении ЭХО-кардиографии обнаружен врожденный порок сердца — открытый аортальный проток (диаметр протока по цветному допплеровскому картированию примерно 2,0 мм), открытое овальное окно (диаметр окна до 3 мм с лево-правым сбросом крови), умеренно расширена легочная артерия. При объективном осмотре в 5 лет: диффузная мышечная гипотония и гипотрофия, сухожильная арефлексия. Слабость аксиальных мышц, голову удерживает плохо, отмечается привычная установка головы с наклоном влево и свисанием вперед, «крыловидные лопатки», слабость лицевой мускулатуры.
Роды третьи, самостоятельные, в срок. При рождении вес 4000 г, длина 53 см, закричал сразу. Домой выписан на 5-е сутки. Голову держит с 1,5 мес, сел самостоятельно в 15 мес, пошел в 18 мес. Первые признаки заболевания появились в 3 мес, когда родители обратили внимание на то, что ребенок стал хуже удерживать голову. При проведении ЭНМГ выявлен первично мышечный уровень поражения. На ЭКГ в 4 года выявлена выраженная синусовая тахикардия, частота сердечных сокращений до 112 ударов в 1 мин. Снижен вольтаж QRS в стандартных и усиленных отведениях. При проведении ЭХО-кардиографии обнаружен врожденный порок сердца — открытый аортальный проток (диаметр протока по цветному допплеровскому картированию примерно 2,0 мм), открытое овальное окно (диаметр окна до 3 мм с лево-правым сбросом крови), умеренно расширена легочная артерия. При объективном осмотре в 5 лет: диффузная мышечная гипотония и гипотрофия, сухожильная арефлексия. Слабость аксиальных мышц, голову удерживает плохо, отмечается привычная установка головы с наклоном влево и свисанием вперед, «крыловидные лопатки», слабость лицевой мускулатуры. Небный и глоточный рефлексы несколько снижены. Отмечается выраженная ригидность спины и шеи, выраженный гиперлордоз поясничного отдела позвоночника, фиксированные контрактуры в тазобедренных и локтевых, начинающиеся — в коленных и голеностопных (больше слева) суставах (рис. 2). В настоящее время может стоять с опорой и пройти с поддержкой до 1 м. В 2014 г. по желанию родителей мальчик обследован во Франции, где обнаружена мутация c.1361Т>А в гене LMNA в гетерозиготном состоянии, приводящая к аминокислотной замене лейцина (Leu) на глутаминовую кислоту (Gln) в 454 положении полипептидной цепи. Эта мутация выявлена впервые.
Небный и глоточный рефлексы несколько снижены. Отмечается выраженная ригидность спины и шеи, выраженный гиперлордоз поясничного отдела позвоночника, фиксированные контрактуры в тазобедренных и локтевых, начинающиеся — в коленных и голеностопных (больше слева) суставах (рис. 2). В настоящее время может стоять с опорой и пройти с поддержкой до 1 м. В 2014 г. по желанию родителей мальчик обследован во Франции, где обнаружена мутация c.1361Т>А в гене LMNA в гетерозиготном состоянии, приводящая к аминокислотной замене лейцина (Leu) на глутаминовую кислоту (Gln) в 454 положении полипептидной цепи. Эта мутация выявлена впервые.
Рис. 2. Больной Б-н, 5 лет. Вид сидя (а) и стоя (б).
Таким образом, нами показано, что на долю ВМД, обусловленной мутациями в гене LMNA, в российской популяции приходится не менее 12% всех случаев этой группы заболеваний. Также полученные результаты свидетельствуют в пользу наличия мажорной мутации в гене LMNA c. 94_96delAAG, которая выявлена у 4 из 5 диагностированных больных.
94_96delAAG, которая выявлена у 4 из 5 диагностированных больных.
Фенотипические проявления этой формы ВМД имеют специфические особенности и характеризуются симптомами прогрессирующего вялого пареза с преимущественным поражением мышц аксиальных и ответственных за подошвенное сгибание стопы. Заболевание манифестирует в интервале от периода новорожденности до грудного возраста. В клинической картине на фоне диффузной гипотрофии и гипотонии отмечается раннее формирование симптома «свисающей головы», контрактур в дистальных отделах нижних конечностей. По мере прогрессирования заболевания у больных формируются контрактуры в других крупных суставах и выраженный гиперлордоз поясничного отдела позвоночника, сохраняющийся даже в положении лежа на спине. В большинстве случаев отмечается заинтересованность лицевой и дыхательной мускулатуры.
Ограниченность выборки не позволяет сделать однозначное заключение, но мы можем согласиться с мнением других авторов, которые указывают на существование двух фенотипов в этой группе ВМД.
Содержание страницы:
Перевод материалов Muscular Dystrophy Association. Ссылка на источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Ссылка на источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Врождённая мышечная дистрофия относится к группе мышечных дистрофий, которые становятся заметными при рождении или сразу после рождения.
Мышечные дистрофии являются, в основном, генетическими дегенеративными заболеваниями, поражающие большей частью произвольно сокращающиеся мышцы.
Дети с врождённой мышечной дистрофией — слабые уже при рождении и могут испытывать трудности с дыханием и глотанием.
В настоящее время методы поддержки больных с ВМД значительно улучшились и повысили выживаемость пациентов, а клинические испытания препаратов против этой болезни — уже не в таком далёком будущем, как это было раньше.
Более подробно см. на mda.org: Types of CMD.
Симптомы ВМДВМД приводит к общей мышечной слабости с возможным развитием суставных контрактур, также возможно ослабление суставов.
В зависимости от типа при ВМД может появиться искривление позвоночника, дыхательная недостаточность, ментальные нарушения, сложности с обучением, дефекты глаз или судороги.
Более подробно см. на mda.org: Types of CMD and Signs and Symptoms.
Причины появления ВМДВМД возникает из-за мутаций в генах, отвечающих за определённые белки, необходимые для мышц, а в некоторых случаях — для глаз или мозга.
Более подробно см. на mda.org: Causes/Inheritance.
Прогрессирование ВМДВМД проявляется при рождении или через короткое время после него. Прогрессирование заболевание зависит от типа. Многие типы прогрессируют медленно, но некоторые приводят к сокращению продолжительности жизни.
Статус исследований по ВМДИсследователи определили многие из генов, дефекты в которых приводят к появлению ВМД. Эти открытия ведут нас к лучшему пониманию природы болезни и улучшают диагностику, а также стратегии лечения.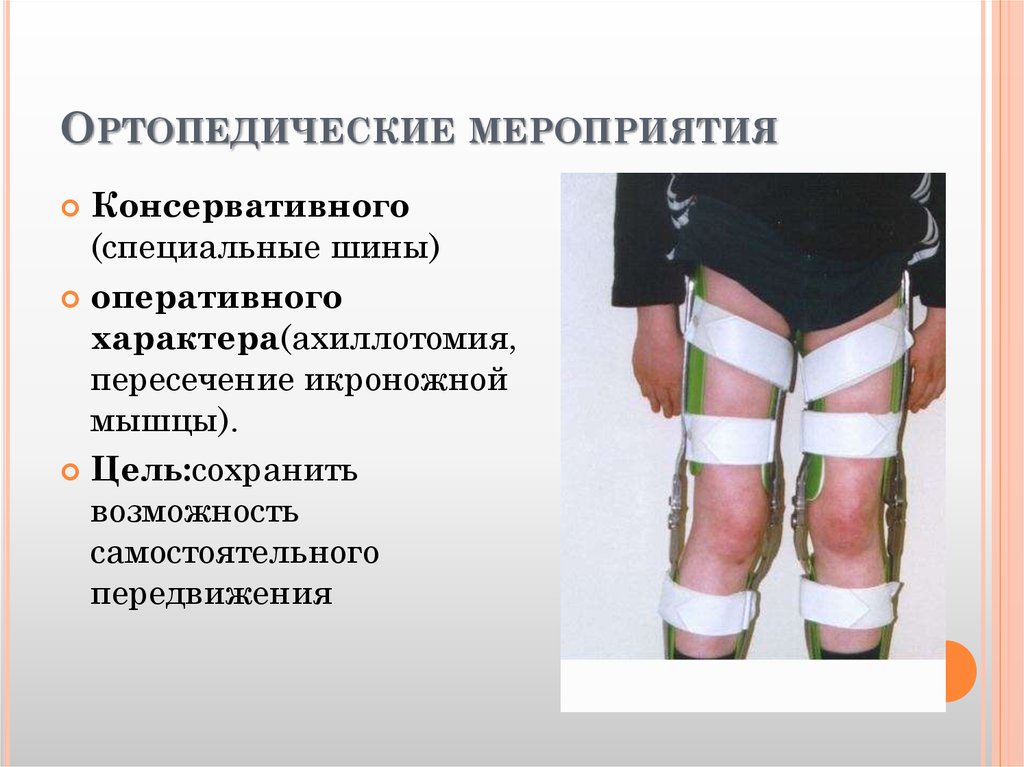
Более подробно см. на mda.org: Research.
Источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Общий список клиник и врачей, занимающихся нервно-мышечными заболеваниями, — по ссылке.
[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-RnIzMDVLNTFCZ2M/preview?usp=drivesdk» title=»2014_Диагностика врожденных МД_Журнал НМБ.pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
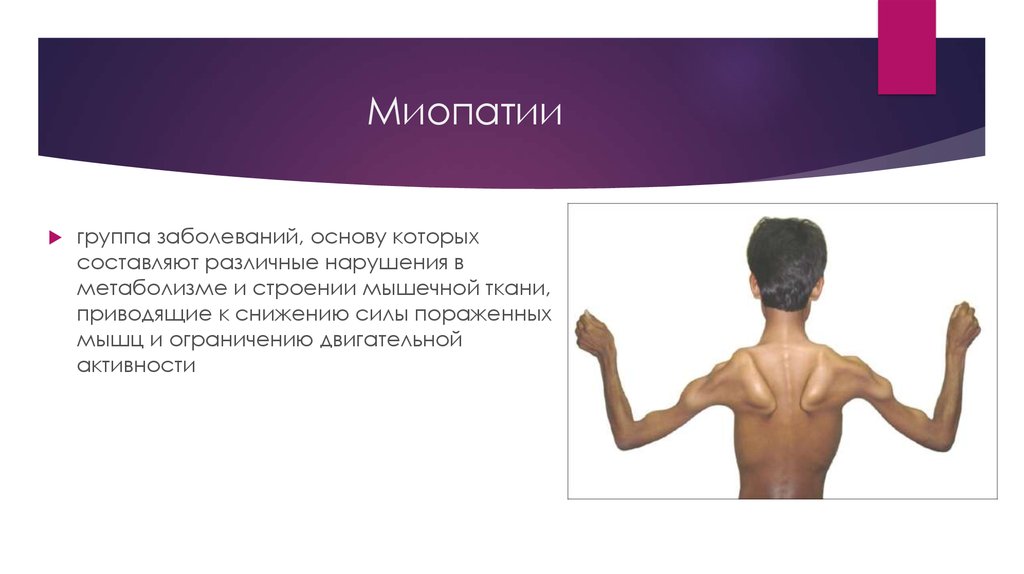 Авторы: Сухоруков В. С., Харламов Д. А. Если статья не видна во врезке ниже, вы можете ознакомиться с текстом на сайте Киберленинки: http://cyberleninka.ru/article/n/differentsialnaya-diagnostika-vrozhdennyh-miopatiy.
Авторы: Сухоруков В. С., Харламов Д. А. Если статья не видна во врезке ниже, вы можете ознакомиться с текстом на сайте Киберленинки: http://cyberleninka.ru/article/n/differentsialnaya-diagnostika-vrozhdennyh-miopatiy.[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-cUhobHF4LUNRMmc/preview?usp=drivesdk» title=»2011_Диагностика врожденных миопатий_Журнал НМБ.pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-dTNOVlF1N1pmN0U/preview?usp=drivesdk» title=»2017-03-31_Семейное-руководство-врождённые миопатии. pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
[embeddoc url=»https://drive.google.com/file/d/0B0m38yuXU_b-TmlyT3diRXotSDA/preview?usp=drive_web» viewer=»drive» ]
[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-b2J4ZkQxZUgwdDg/preview?usp=drivesdk» title=»2017-03-31_Для врачей_Стандарты лечения врождённых миопатий.docx» icon=»https://drive-thirdparty. googleusercontent.com/16/type/application/vnd.openxmlformats-officedocument.wordprocessingml.document» width=»100%» height=»400″ style=»embed»]
googleusercontent.com/16/type/application/vnd.openxmlformats-officedocument.wordprocessingml.document» width=»100%» height=»400″ style=»embed»]
Регистры пациентов в разных странах вы можете узнать по ссылке: http://www.treat-nmd.eu/resources/patient-registries/list/CMD/.
Международный регистр по врождённым мышечным заболеваниям (CMDIR — Congenital muscle disease international register).
Почему нужно регистрироваться в реестре?По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку генетические нервно-мышечные заболевания являются редкими (орфанными) заболеваниями.
Для этого в разных странах ведутся реестры пациентов — базы данных по генетической и клинической информации о людях, страдающих от генетических нервно-мышечных заболеваний и желающих ускорить процесс исследований.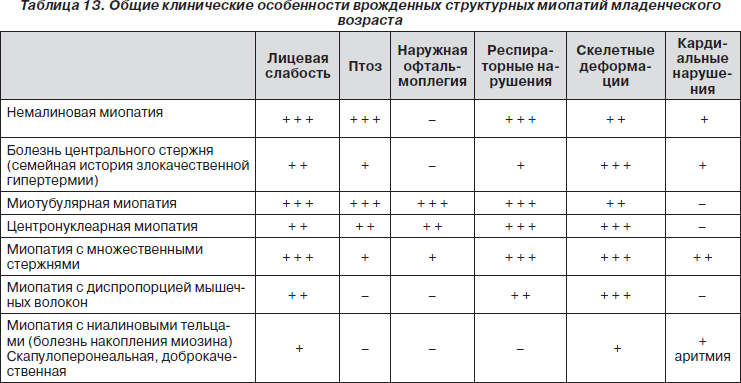 Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Статьи о ВМД на нашем сайте
Статьи о ВМД типа 1А на нашем сайте
Divij Pasrija; Прасанна Тади.
Информация об авторе
Последнее обновление: 4 июля 2022 г.
Врожденная мышечная дистрофия — один из вариантов нарушений мышечной слабости, проявляющихся в раннем возрасте, в младенчестве и вскоре после рождения. Разница между врожденными миопатиями и мышечными дистрофиями заключается в том, что дистрофии постепенно прогрессируют и связаны с увеличением разрушения мышц с возрастом. Врожденные миопатии означают нарушения, связанные со слабостью мышц в неонатальной возрастной группе, которые могут быть вторичными по отношению к генетическим, метаболическим или другим нарушениям и обычно имеют непрогрессирующее течение. В этом мероприятии рассматривается классификация, оценка и лечение врожденных мышечных дистрофий и подчеркивается роль межпрофессиональной команды в оценке и лечении пациентов с этим заболеванием.
Цели:
Определите различные причины мышечной слабости, проявляющейся в раннем детстве.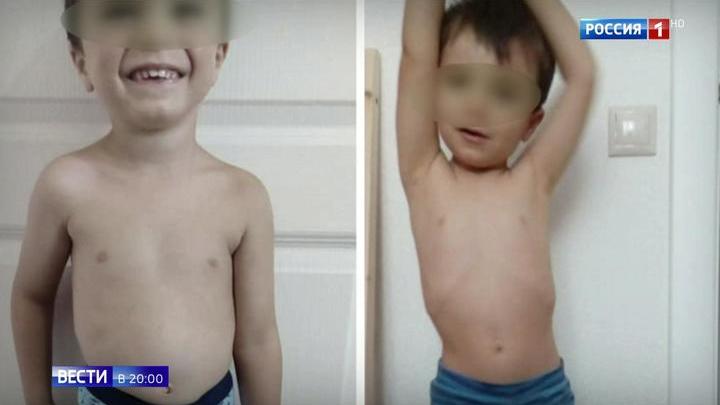
Опишите различные категории мышечных дистрофий на основе задействованных генетических моделей.
Кратко о диагностике и лечении мышечных дистрофий.
Опишите роль различных узких специалистов и поставщиков медицинских услуг в ведении этих пациентов для улучшения качества жизни и результатов.
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Врожденная мышечная дистрофия является одним из вариантов нарушений мышечной слабости, проявляющихся в раннем возрасте, в младенчестве и вскоре после рождения. Разница между врожденными миопатиями и мышечными дистрофиями заключается в том, что дистрофии постепенно прогрессируют и связаны с повышенным разрушением мышц с возрастом.[1] Врожденные миопатии означают нарушения, связанные со слабостью мышц в неонатальной возрастной группе, которые могут быть вторичными по отношению к генетическим, метаболическим или другим нарушениям и обычно имеют непрогрессирующее течение. Классификация этих групп заболеваний основана на результатах биопсии мышц и генетической оценке. Общими для этих заболеваний данными биопсии мышц являются атрофия мышечных волокон, фиброзно-жировая инфильтрация ткани.
Классификация этих групп заболеваний основана на результатах биопсии мышц и генетической оценке. Общими для этих заболеваний данными биопсии мышц являются атрофия мышечных волокон, фиброзно-жировая инфильтрация ткани.
Большинство этих заболеваний наследуются и связаны со специфическими генами. Они влияют на синтез различных белков на границе внеклеточного матрикса плазматической мембраны. Наиболее частый тип наследования – аутосомно-рецессивный. Болезнь Дюшенна сцеплена с Х-хромосомой и поэтому встречается только у мальчиков. Большинство самок являются носителями, но у некоторых может наблюдаться мышечная слабость от легкой до умеренной степени. Как дистрофия Дюшенна, так и дистрофия Беккера являются результатом мутации гена дистрофина, локус которого - Xp21.2. Дистрофия Дюшенна возникает, когда белок аномально усечен, тогда как дистрофия Беккера возникает из-за частично функционального дистрофина.
Другие врожденные дистрофии, включая дистрофию Уокера Варбурга, мышечно-глазной мозг и болезнь Фукуямы, вместе называются а-дистрогликанопатиями (из-за мутации, наблюдаемой в общем белке а-дистрогликана). Миотоническая дистрофия обычно передается по аутосомно-доминантному типу и возникает в результате экспансии тринуклеотидных (CTG) повторов в гене DMPK в локусе 19q13.3.[3] Миопатия Ульриха и Вифлеема возникает в результате изменения молекул коллагена VI в трех известных генах, COL6A1, COL6A2 и COL6A3, и может быть аутосомно-рецессивным или доминантным.
Миотоническая дистрофия обычно передается по аутосомно-доминантному типу и возникает в результате экспансии тринуклеотидных (CTG) повторов в гене DMPK в локусе 19q13.3.[3] Миопатия Ульриха и Вифлеема возникает в результате изменения молекул коллагена VI в трех известных генах, COL6A1, COL6A2 и COL6A3, и может быть аутосомно-рецессивным или доминантным.
Из всех врожденных мышечных дистрофий мышечная дистрофия Дюшенна является наиболее распространенной, и ее частота составляет около 1 на 3600 мальчиков.[4] Заболеваемость врожденными мышечными дистрофиями у детей в популяционных исследованиях оценивается примерно в 0,82/100 000 детей.[5] Преобладание определенных типов также может быть обычным явлением в зависимости от географического региона, например, мышечная дистрофия Фукуямы является наиболее распространенным типом в Японии.
Делеция или мальформация определенных белков, вторичная по отношению к унаследованным мутациям или мутациям de novo, приводит к аномальной структуре или функции белка и, таким образом, к мышечной слабости.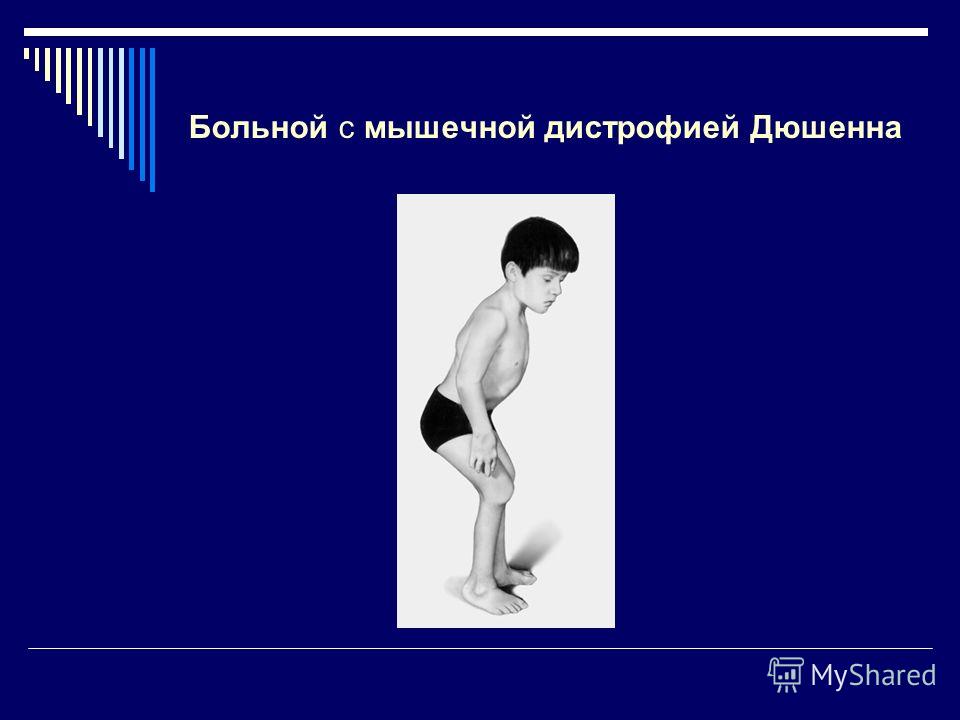 Распространенные формы врожденных мышечных дистрофий возникают в результате изменений в поверхностных белках плазматической мембраны или в белках, образующих часть поверхности раздела мембрана-внеклеточный матрикс.[7] Открытие мерозина, белка внеклеточного матрикса, привело к классификации заболевания на мерозин-положительные и мерозин-отрицательные формы. Мерозин-отрицательная форма была связана с выраженной двигательной недостаточностью, высокими уровнями креатинкиназы и относительно нормальными показателями IQ.[8] Тем не менее, пациенты с отрицательным мерозином были связаны с аномальными результатами МРТ белого вещества с сохранением базальных ганглиев.]
Распространенные формы врожденных мышечных дистрофий возникают в результате изменений в поверхностных белках плазматической мембраны или в белках, образующих часть поверхности раздела мембрана-внеклеточный матрикс.[7] Открытие мерозина, белка внеклеточного матрикса, привело к классификации заболевания на мерозин-положительные и мерозин-отрицательные формы. Мерозин-отрицательная форма была связана с выраженной двигательной недостаточностью, высокими уровнями креатинкиназы и относительно нормальными показателями IQ.[8] Тем не менее, пациенты с отрицательным мерозином были связаны с аномальными результатами МРТ белого вещества с сохранением базальных ганглиев.]
Основные категории врожденных мышечных дистрофий, основанные на пораженных белках: мутация в белке ламинин альфа 2, который является дефицитом мерозина[11]
Связанный с альфа-дистрогликаном – включая ходока Варбурга, мышечно-глазной мозг и миопатию Фукуямы (ген Фукутина в 9q31-33)[12] и дистрофия поясно-конечностных мышц
Соотношение между генотипом и фенотипом зависит от того, является ли пораженный белок полностью или частично дефицитным, что определяется типом мутации в гене.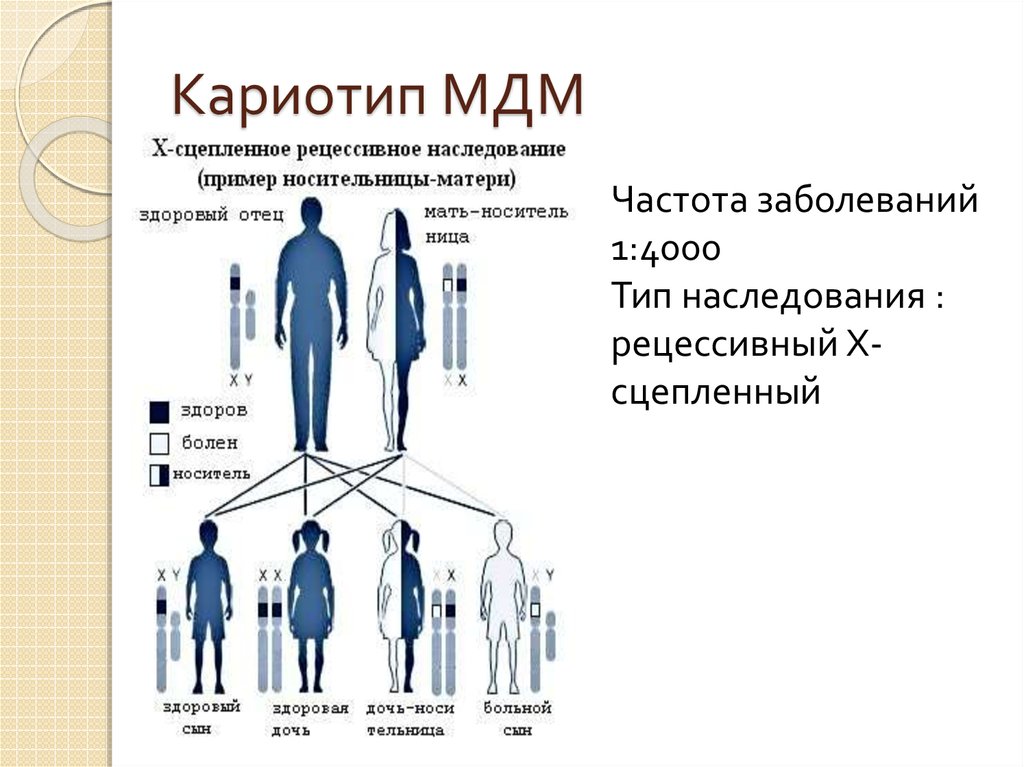
При антенатальной беременности пациенток, страдающих мышечной дистрофией, может наблюдаться многоводие из-за уменьшения проглатывания амниотической жидкости и уменьшения движений плода в утробе матери. При рождении у этих пациентов может наблюдаться слабый слабый крик и минимальные спонтанные движения конечностей, гипотония при осмотре, а в наиболее тяжелых вариантах могут быть контрактуры при рождении [13].
При опросе родителей можно выяснить, что в анамнезе плохое питание, вторичное по отношению к плохому сосанию. Значительная задержка моторики может быть одним из других симптомов, проявляющихся в детстве. Тяжесть мышечной слабости у этих пациентов зависит от типа заболевания и полного или частичного поражения аномального белка. Всех младенцев с подозрением на гипотонию следует тщательно обследовать, чтобы исключить аномалии, поражающие другие органы, и выявить возможную синдромальную связь.[14]
Время начала (младенчество или детство) и тип задействованных мышц (проксимальные или дистальные) являются ключом к ограничению дифференциальной диагностики меньшим набором расстройств.
Скрининговое обследование младенца с гипотонией должно включать:
Невролог должен провести тщательный анамнез и клинический осмотр, документирующий мощность каждого сустава и рефлексы.
Следует учитывать уровень креатинкиназы (КК), альдолазы, аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ), исследования нервной проводимости и ЭМГ. Однако уровни креатинкиназы могут варьироваться от полностью нормальных до значительно повышенных в зависимости от фенотипа. Повышенный уровень КК, альдолазы обычно свидетельствует о дистрофическом процессе.
Биопсия мышц, которая ранее выполнялась в плановом порядке как часть оценки этих заболеваний, больше не используется во всех случаях с появлением геномного секвенирования. Иммуногистохимия и электронная микроскопия могут помочь идентифицировать специфические заболевания на основе моделей окрашивания.
Эхокардиография и ЭКГ должны быть выполнены для выявления кардиомиопатии и аномальной проводимости, которые обычно возникают при этих заболеваниях, если аномальный белок также экспрессируется в сердечных миоцитах.
По возможности следует выполнять МРТ головного мозга, так как многие из этих нарушений связаны со специфическими аномалиями в различных областях мозга на последовательностях визуализации.
Определенные заболевания, такие как дистрофия Фукуямы и мышечно-глазная болезнь головного мозга, связаны с патологиями глаз, такими как близорукость, косоглазие, катаракта и глаукома, у значительной части пациентов.
Подтверждающее тестирование для большинства из этих заболеваний требует тестирования на конкретный белок или генетические изменения или мутации, которые основаны на основном заболевании, и предпочтительно должно проводиться в лаборатории, специализирующейся на проведении генетического тестирования.
Ведение большинства из этих заболеваний является поддерживающим, поскольку замена определенных белков и генов была опробована в основном на животных моделях. Использование этих лекарств и генной терапии на людях было частью клинических испытаний.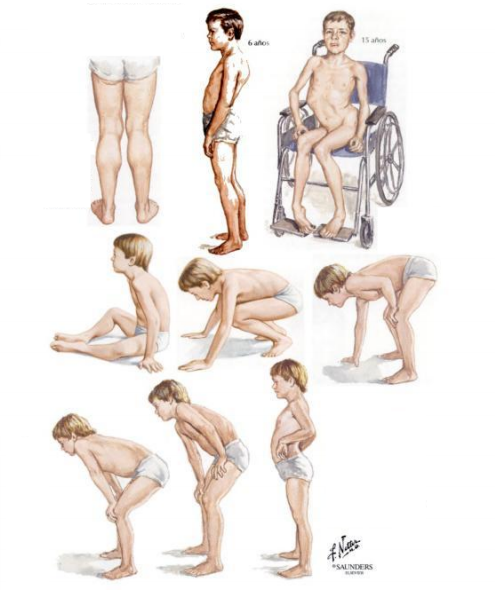 [15]
[15]
В настоящее время в большинстве центров, занимающихся лечением таких пациентов, есть межпрофессиональные бригады, в состав которых входит несколько узких специалистов, которые помогают лицам, осуществляющим уход, ухаживать за такими пациентами с многочисленными сложными потребностями на дому.
Вспомогательная искусственная вентиляция легких проводится дома с помощью родителей и патронажа при поражении дыхательных мышц.
Интенсивная физиотерапия и трудотерапия являются неотъемлемой частью ухода за такими пациентами во время госпитализации и должны продолжаться дома.
Эти пациенты нуждаются в хирургическом вмешательстве по поводу других сопутствующих заболеваний, таких как сколиоз из-за длительной неподвижности, установки питающей гастростомы или гастроеюноанастомоза для удовлетворения потребностей в питании.[16]
Нутритивная поддержка играет важную роль в долгосрочном ведении этих пациентов, поскольку они обычно имеют недостаточный вес, а дефицит макро-/микроэлементов у них может усугубить лежащую в основе мышечную слабость.
Многие пациенты могут иметь нормальные интеллектуальные способности и поэтому нуждаются в поддержке в виде психологов и психиатров, чтобы помочь им справиться со стрессом хронического заболевания.
Дифференциальный диагноз пациентов со слабостью в раннем младенчестве включает:
Врожденные миопатии – наиболее распространенными являются центральное ядро, немалиновая дорога и центроядерная миопатия. Они также могут быть вторичными по отношению к метаболическим нарушениям, влияющим на метаболизм углеводов, жиров и других молекул.
Заболевания мионеврального синапса – встречаются очень редко, но должны быть частью дифференциальной диагностики младенцев со слабостью. К ним относятся врожденная миастения и младенческий ботулизм.
Нейропатии – это заболевания, поражающие преимущественно нервные корешки спинного мозга и периферические нервы. Общие невропатии, проявляющиеся в раннем детстве, включают спинальную мышечную атрофию (СМА) и наследственную моторно-сенсорную невропатию (HMSN).
Поскольку лечение большинства этих заболеваний является поддерживающим, долгосрочный прогноз плохой, а ожидаемая продолжительность жизни зависит от корреляции между лежащим в основе генетическим дефектом и фенотипом, вторичным по отношению к аномальному белку. Наиболее частая причина заболеваемости и смертности при этих заболеваниях вторична по отношению к респираторным или сердечным осложнениям из-за мышечной слабости.
Респиратор
Хроническая дыхательная недостаточность, вторичная по отношению к мышечной слабости, часто приводит к ателектазам, аспирации и пневмонии, вторичной по отношению к ИВЛ.
Сердечный
Кардиомиопатия может быть распространенным сопутствующим заболеванием, и у них может быть хроническая сердечная недостаточность, которая может потребовать медикаментозного лечения.
Опорно-двигательный аппарат
У них может быть сколиоз из-за неподвижности, остеопении и повышенного риска переломов. Также могут развиваться пролежни и контрактуры суставов, даже несмотря на агрессивную физиотерапию.
Также могут развиваться пролежни и контрактуры суставов, даже несмотря на агрессивную физиотерапию.
Эндокринный
У этих пациентов часто встречается надпочечниковая недостаточность, и в случае стресса им может потребоваться прием стероидов. Уровни щитовидной железы и витамина D также следует оценивать в рамках комплексной оценки.
Неврологический
У некоторых из этих пациентов могут быть судороги; Бремя психологических расстройств, таких как депрессия и тревога, у этих пациентов велико из-за основного хронического заболевания.
Ранняя диагностика является ключом к лечению этих расстройств, и при наличии предшествующего семейного анамнеза можно рассмотреть возможность проведения дородового тестирования с анализом ДНК беременной женщины.
Невролог должен осмотреть любого младенца с необъяснимой мышечной слабостью, а генетическое тестирование должно быть частью комплексной оценки, поскольку многие из этих расстройств могут иметь перекрывающиеся признаки.
Установление подтвержденного генетического диагноза необходимо для прогнозирования, консультирования и пренатального тестирования будущих беременностей.
Вскоре после постановки диагноза к уходу за такими пациентами должна быть привлечена межпрофессиональная бригада, поскольку у них могут быть множественные медицинские и психосоциальные проблемы. Лица, осуществляющие уход, должны быть хорошо обучены уходу за медицинским оборудованием, которое требуется этим пациентам дома, таким как вентиляторы и трубки для кормления. Для быстрого решения любых основных проблем требуется регулярное последующее наблюдение, и это может улучшить качество жизни этих пациентов.
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Комментарий к этой статье.
Floriach-Robert M, Cabello A, Simon De Las Heras R, Mateos Beato F. [Неонатальная гипотония мышечного происхождения: анализ 50 случаев]. Неврология. 2001 июнь-июль;16(6):245-53. [PubMed: 11423041]
[Неонатальная гипотония мышечного происхождения: анализ 50 случаев]. Неврология. 2001 июнь-июль;16(6):245-53. [PubMed: 11423041]
Ю Э.М., Корнберг А.Дж. Мышечная дистрофия Дюшенна. J Педиатр Здоровье ребенка. 2015 авг; 51 (8): 759-64. [PubMed: 25752877]
Zapata-Aldana E, Ceballos-Sáenz D, Hicks R, Campbell C. Пренатальные, неонатальные и ранние детские особенности врожденной миотонической дистрофии. J нервно-мышечной Dis. 2018;5(3):331-340. [PubMed: 30010141]
Ши ПБ. Мышечные дистрофии и другие генетические миопатии. Нейрол клин. 2013 ноябрь;31(4):1009-29. [PubMed: 24176421]
Mah JK, Korngut L, Fiest KM, Dykeman J, Day LJ, Pringsheim T, Jette N. Систематический обзор и метаанализ эпидемиологии мышечных дистрофий. Может J Neurol Sci. 2016 Январь; 43 (1): 163-77. [В паблике: 26786644]
Окада М., Кавахара Г., Ногучи С., Суги К.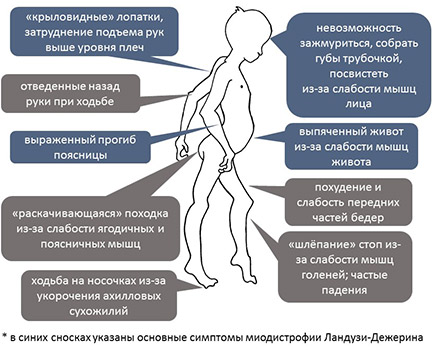 , Мураяма К., Нонака И., Хаяши Ю.К., Нишино И. Первичный дефицит коллагена VI является второй наиболее распространенной врожденной мышечной дистрофией в Японии. Неврология. 04 сентября 2007 г .; 69 (10): 1035-42. [PubMed: 17785673]
, Мураяма К., Нонака И., Хаяши Ю.К., Нишино И. Первичный дефицит коллагена VI является второй наиболее распространенной врожденной мышечной дистрофией в Японии. Неврология. 04 сентября 2007 г .; 69 (10): 1035-42. [PubMed: 17785673]
Schorling DC, Kirschner J, Bönnemann CG. Врожденные мышечные дистрофии и миопатии: обзор и обновление. нейропедиатрия. 2017 авг; 48 (4): 247-261. [PubMed: 28669131]
Кобаяши О., Хаяши Ю., Арахата К., Одзава Э., Нонака И. Врожденная мышечная дистрофия: клиническое и патологическое исследование 50 пациентов с классической (западной) мерозин-положительной формой. Неврология. 1996 март; 46 (3): 815-8. [PubMed: 8618689]
Philpot J, Sewry C, Pennock J, Dubowitz V. Клинический фенотип при врожденной мышечной дистрофии: корреляция с экспрессией мерозина в скелетных мышцах. Нервно-мышечное расстройство. 1995 г., июль; 5(4):301-5. [В паблике: 7580243]

Bönnemann CG. Миопатии, связанные с коллагеном VI, врожденная мышечная дистрофия Ульриха и миопатия Бетлема. Handb Clin Neurol. 2011;101:81-96. [Бесплатная статья PMC: PMC5207779] [PubMed: 21496625]
Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé FM, Schwartz K, Fardeau M, Tryggvason K. Мутации в гене 2-цепи ламинина альфа (LAMA2) вызывают врожденную мышечную дистрофию с дефицитом мерозина. Нат Жене. 1995 октября; 11 (2): 216-8. [PubMed: 7550355]
Falsaperla R, Praticò AD, Ruggieri M, Parano E, Rizzo R, Corsello G, Vitaliti G, Pavone P. Врожденная мышечная дистрофия: от мышц к мозгу. Ital J Pediatr. 2016 31 августа; 42(1):78. [Бесплатная статья PMC: PMC5006267] [PubMed: 27576556]
Исигаки К., Ихара С., Накамура Х., Мори-Йошимура М., Маруо К., Танигучи-Икеда М., Кимура Э., Мураками Т., Сато Т. , Тода Т., Кайя Х., Осава М. Национальный реестр пациентов с врожденной мышечной дистрофией Фукуямы в Японии. Нервно-мышечное расстройство. 2018 Октябрь; 28 (10): 885-893. [PubMed: 30220444]
Нервно-мышечное расстройство. 2018 Октябрь; 28 (10): 885-893. [PubMed: 30220444]
Спаркс ЮВ. Неонатальная гипотония. Клин Перинатол. 2015 июнь;42(2):363-71, ix. [PubMed: 26042909]
Барраса-Флорес П., Бейтс Ч.Р., Оливейра-Сантос А., Буркин Д.Дж. Ламинин и интегрин при врожденной мышечной дистрофии, связанной с LAMA2: от болезни к терапии. Фронт Мол Невроски. 2020;13:1. [Бесплатная статья PMC: PMC7026472] [PubMed: 32116540]
Баласубраманиан М., Сэйерс Р., Мартиндейл Дж. Врожденная миотоническая дистрофия: естественное прогрессирование заболевания и лицевая дисморфология. Клин Дисморфол. 2014 Октябрь; 23 (4): 127-9. [PubMed: 25144154]
Divij Pasrija; Прасанна Тади.
Информация об авторе
Последнее обновление: 4 июля 2022 г.
Врожденная мышечная дистрофия — один из вариантов нарушений мышечной слабости, проявляющихся в раннем возрасте, в младенчестве и вскоре после рождения.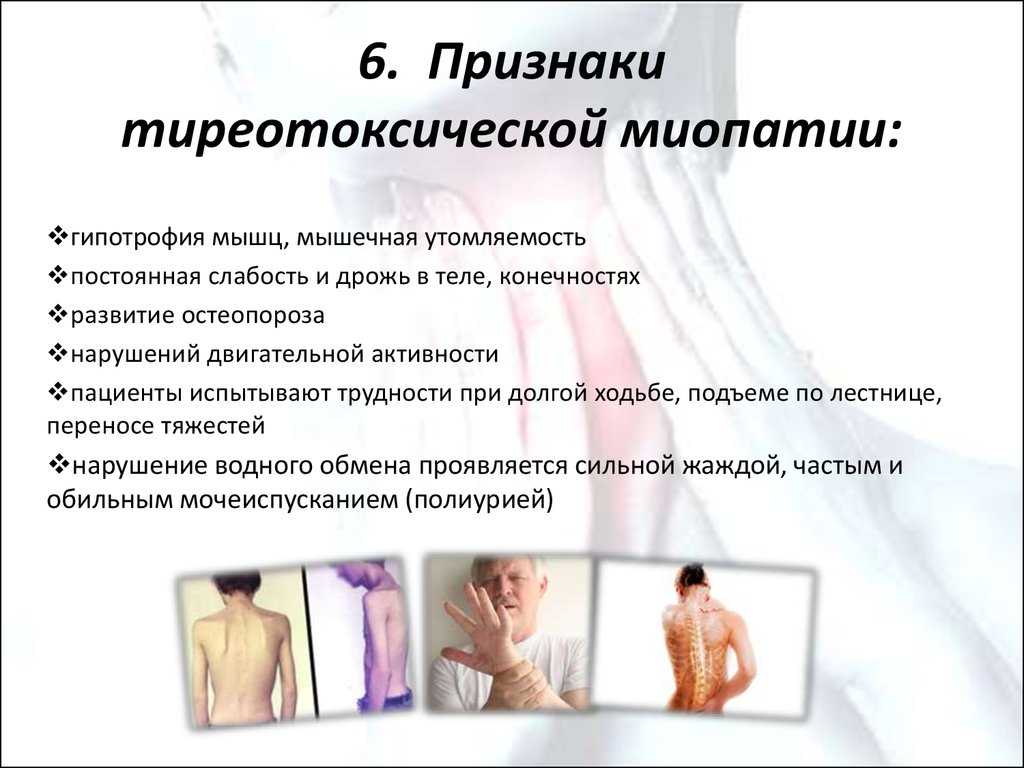 Разница между врожденными миопатиями и мышечными дистрофиями заключается в том, что дистрофии постепенно прогрессируют и связаны с увеличением разрушения мышц с возрастом. Врожденные миопатии означают нарушения, связанные со слабостью мышц в неонатальной возрастной группе, которые могут быть вторичными по отношению к генетическим, метаболическим или другим нарушениям и обычно имеют непрогрессирующее течение. В этом мероприятии рассматривается классификация, оценка и лечение врожденных мышечных дистрофий и подчеркивается роль межпрофессиональной команды в оценке и лечении пациентов с этим заболеванием.
Разница между врожденными миопатиями и мышечными дистрофиями заключается в том, что дистрофии постепенно прогрессируют и связаны с увеличением разрушения мышц с возрастом. Врожденные миопатии означают нарушения, связанные со слабостью мышц в неонатальной возрастной группе, которые могут быть вторичными по отношению к генетическим, метаболическим или другим нарушениям и обычно имеют непрогрессирующее течение. В этом мероприятии рассматривается классификация, оценка и лечение врожденных мышечных дистрофий и подчеркивается роль межпрофессиональной команды в оценке и лечении пациентов с этим заболеванием.
Цели:
Определите различные причины мышечной слабости, проявляющейся в раннем детстве.
Опишите различные категории мышечных дистрофий на основе задействованных генетических моделей.
Кратко о диагностике и лечении мышечных дистрофий.
Опишите роль различных узких специалистов и поставщиков медицинских услуг в ведении этих пациентов для улучшения качества жизни и результатов.
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Врожденная мышечная дистрофия является одним из вариантов нарушений мышечной слабости, проявляющихся в раннем возрасте, в младенчестве и вскоре после рождения. Разница между врожденными миопатиями и мышечными дистрофиями заключается в том, что дистрофии постепенно прогрессируют и связаны с повышенным разрушением мышц с возрастом.[1] Врожденные миопатии означают нарушения, связанные со слабостью мышц в неонатальной возрастной группе, которые могут быть вторичными по отношению к генетическим, метаболическим или другим нарушениям и обычно имеют непрогрессирующее течение. Классификация этих групп заболеваний основана на результатах биопсии мышц и генетической оценке. Общими для этих заболеваний данными биопсии мышц являются атрофия мышечных волокон, фиброзно-жировая инфильтрация ткани.
Большинство этих заболеваний наследуются и связаны со специфическими генами.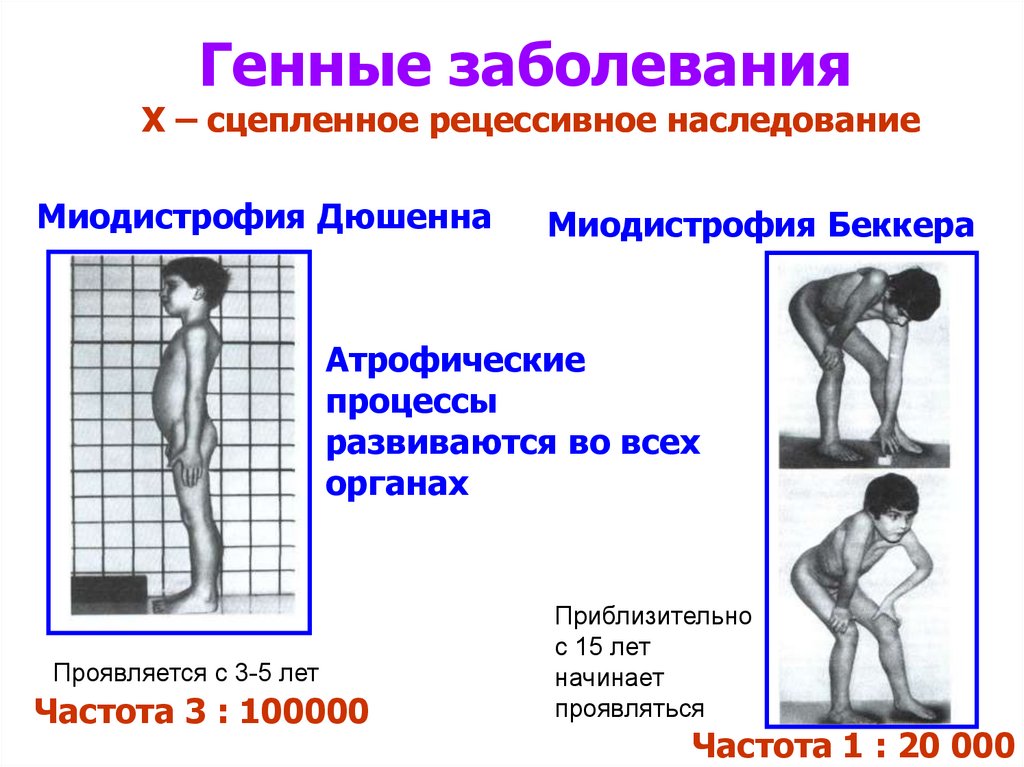 Они влияют на синтез различных белков на границе внеклеточного матрикса плазматической мембраны. Наиболее частый тип наследования – аутосомно-рецессивный. Болезнь Дюшенна сцеплена с Х-хромосомой и поэтому встречается только у мальчиков. Большинство самок являются носителями, но у некоторых может наблюдаться мышечная слабость от легкой до умеренной степени. Как дистрофия Дюшенна, так и дистрофия Беккера являются результатом мутации гена дистрофина, локус которого - Xp21.2. Дистрофия Дюшенна возникает, когда белок аномально усечен, тогда как дистрофия Беккера возникает из-за частично функционального дистрофина.
Они влияют на синтез различных белков на границе внеклеточного матрикса плазматической мембраны. Наиболее частый тип наследования – аутосомно-рецессивный. Болезнь Дюшенна сцеплена с Х-хромосомой и поэтому встречается только у мальчиков. Большинство самок являются носителями, но у некоторых может наблюдаться мышечная слабость от легкой до умеренной степени. Как дистрофия Дюшенна, так и дистрофия Беккера являются результатом мутации гена дистрофина, локус которого - Xp21.2. Дистрофия Дюшенна возникает, когда белок аномально усечен, тогда как дистрофия Беккера возникает из-за частично функционального дистрофина.
Другие врожденные дистрофии, включая дистрофию Уокера Варбурга, мышечно-глазной мозг и болезнь Фукуямы, вместе называются а-дистрогликанопатиями (из-за мутации, наблюдаемой в общем белке а-дистрогликана). Миотоническая дистрофия обычно передается по аутосомно-доминантному типу и возникает в результате экспансии тринуклеотидных (CTG) повторов в гене DMPK в локусе 19q13.3.[3] Миопатия Ульриха и Вифлеема возникает в результате изменения молекул коллагена VI в трех известных генах, COL6A1, COL6A2 и COL6A3, и может быть аутосомно-рецессивным или доминантным.
Из всех врожденных мышечных дистрофий мышечная дистрофия Дюшенна является наиболее распространенной, и ее частота составляет около 1 на 3600 мальчиков.[4] Заболеваемость врожденными мышечными дистрофиями у детей в популяционных исследованиях оценивается примерно в 0,82/100 000 детей.[5] Преобладание определенных типов также может быть обычным явлением в зависимости от географического региона, например, мышечная дистрофия Фукуямы является наиболее распространенным типом в Японии.
Делеция или мальформация определенных белков, вторичная по отношению к унаследованным мутациям или мутациям de novo, приводит к аномальной структуре или функции белка и, таким образом, к мышечной слабости. Распространенные формы врожденных мышечных дистрофий возникают в результате изменений в поверхностных белках плазматической мембраны или в белках, образующих часть поверхности раздела мембрана-внеклеточный матрикс.[7] Открытие мерозина, белка внеклеточного матрикса, привело к классификации заболевания на мерозин-положительные и мерозин-отрицательные формы.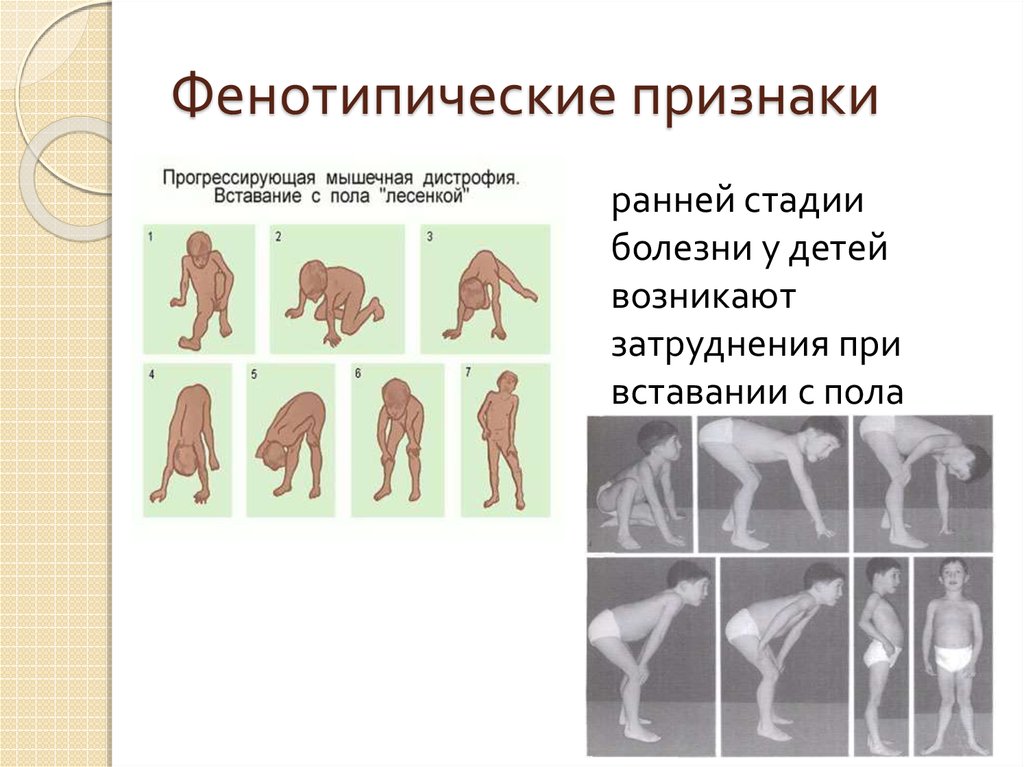 Мерозин-отрицательная форма была связана с выраженной двигательной недостаточностью, высокими уровнями креатинкиназы и относительно нормальными показателями IQ.[8] Тем не менее, пациенты с отрицательным мерозином были связаны с аномальными результатами МРТ белого вещества с сохранением базальных ганглиев.]
Мерозин-отрицательная форма была связана с выраженной двигательной недостаточностью, высокими уровнями креатинкиназы и относительно нормальными показателями IQ.[8] Тем не менее, пациенты с отрицательным мерозином были связаны с аномальными результатами МРТ белого вещества с сохранением базальных ганглиев.]
Основные категории врожденных мышечных дистрофий, основанные на пораженных белках: мутация в белке ламинин альфа 2, который является дефицитом мерозина[11]
Связанный с альфа-дистрогликаном – включая ходока Варбурга, мышечно-глазной мозг и миопатию Фукуямы (ген Фукутина в 9q31-33)[12] и дистрофия поясно-конечностных мышц
Соотношение между генотипом и фенотипом зависит от того, является ли пораженный белок полностью или частично дефицитным, что определяется типом мутации в гене.
При антенатальной беременности пациенток, страдающих мышечной дистрофией, может наблюдаться многоводие из-за уменьшения проглатывания амниотической жидкости и уменьшения движений плода в утробе матери. При рождении у этих пациентов может наблюдаться слабый слабый крик и минимальные спонтанные движения конечностей, гипотония при осмотре, а в наиболее тяжелых вариантах могут быть контрактуры при рождении [13].
При рождении у этих пациентов может наблюдаться слабый слабый крик и минимальные спонтанные движения конечностей, гипотония при осмотре, а в наиболее тяжелых вариантах могут быть контрактуры при рождении [13].
При опросе родителей можно выяснить, что в анамнезе плохое питание, вторичное по отношению к плохому сосанию. Значительная задержка моторики может быть одним из других симптомов, проявляющихся в детстве. Тяжесть мышечной слабости у этих пациентов зависит от типа заболевания и полного или частичного поражения аномального белка. Всех младенцев с подозрением на гипотонию следует тщательно обследовать, чтобы исключить аномалии, поражающие другие органы, и выявить возможную синдромальную связь.[14]
Время начала (младенчество или детство) и тип задействованных мышц (проксимальные или дистальные) являются ключом к ограничению дифференциальной диагностики меньшим набором расстройств.
Скрининговое обследование младенца с гипотонией должно включать:
Невролог должен провести тщательный анамнез и клинический осмотр, документирующий мощность каждого сустава и рефлексы.
Следует учитывать уровень креатинкиназы (КК), альдолазы, аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ), исследования нервной проводимости и ЭМГ. Однако уровни креатинкиназы могут варьироваться от полностью нормальных до значительно повышенных в зависимости от фенотипа. Повышенный уровень КК, альдолазы обычно свидетельствует о дистрофическом процессе.
Биопсия мышц, которая ранее выполнялась в плановом порядке как часть оценки этих заболеваний, больше не используется во всех случаях с появлением геномного секвенирования. Иммуногистохимия и электронная микроскопия могут помочь идентифицировать специфические заболевания на основе моделей окрашивания.
Эхокардиография и ЭКГ должны быть выполнены для выявления кардиомиопатии и аномальной проводимости, которые обычно возникают при этих заболеваниях, если аномальный белок также экспрессируется в сердечных миоцитах.
По возможности следует выполнять МРТ головного мозга, так как многие из этих нарушений связаны со специфическими аномалиями в различных областях мозга на последовательностях визуализации.
Определенные заболевания, такие как дистрофия Фукуямы и мышечно-глазная болезнь головного мозга, связаны с патологиями глаз, такими как близорукость, косоглазие, катаракта и глаукома, у значительной части пациентов.
Подтверждающее тестирование для большинства из этих заболеваний требует тестирования на конкретный белок или генетические изменения или мутации, которые основаны на основном заболевании, и предпочтительно должно проводиться в лаборатории, специализирующейся на проведении генетического тестирования.
Ведение большинства из этих заболеваний является поддерживающим, поскольку замена определенных белков и генов была опробована в основном на животных моделях. Использование этих лекарств и генной терапии на людях было частью клинических испытаний.[15]
В настоящее время в большинстве центров, занимающихся лечением таких пациентов, есть межпрофессиональные бригады, в состав которых входит несколько узких специалистов, которые помогают лицам, осуществляющим уход, ухаживать за такими пациентами с многочисленными сложными потребностями на дому.
Вспомогательная искусственная вентиляция легких проводится дома с помощью родителей и патронажа при поражении дыхательных мышц.
Интенсивная физиотерапия и трудотерапия являются неотъемлемой частью ухода за такими пациентами во время госпитализации и должны продолжаться дома.
Эти пациенты нуждаются в хирургическом вмешательстве по поводу других сопутствующих заболеваний, таких как сколиоз из-за длительной неподвижности, установки питающей гастростомы или гастроеюноанастомоза для удовлетворения потребностей в питании.[16]
Нутритивная поддержка играет важную роль в долгосрочном ведении этих пациентов, поскольку они обычно имеют недостаточный вес, а дефицит макро-/микроэлементов у них может усугубить лежащую в основе мышечную слабость.
Многие пациенты могут иметь нормальные интеллектуальные способности и поэтому нуждаются в поддержке в виде психологов и психиатров, чтобы помочь им справиться со стрессом хронического заболевания.
Дифференциальный диагноз пациентов со слабостью в раннем младенчестве включает:
Врожденные миопатии – наиболее распространенными являются центральное ядро, немалиновая дорога и центроядерная миопатия. Они также могут быть вторичными по отношению к метаболическим нарушениям, влияющим на метаболизм углеводов, жиров и других молекул.
Заболевания мионеврального синапса – встречаются очень редко, но должны быть частью дифференциальной диагностики младенцев со слабостью. К ним относятся врожденная миастения и младенческий ботулизм.
Нейропатии – это заболевания, поражающие преимущественно нервные корешки спинного мозга и периферические нервы. Общие невропатии, проявляющиеся в раннем детстве, включают спинальную мышечную атрофию (СМА) и наследственную моторно-сенсорную невропатию (HMSN).
Поскольку лечение большинства этих заболеваний является поддерживающим, долгосрочный прогноз плохой, а ожидаемая продолжительность жизни зависит от корреляции между лежащим в основе генетическим дефектом и фенотипом, вторичным по отношению к аномальному белку. Наиболее частая причина заболеваемости и смертности при этих заболеваниях вторична по отношению к респираторным или сердечным осложнениям из-за мышечной слабости.
Наиболее частая причина заболеваемости и смертности при этих заболеваниях вторична по отношению к респираторным или сердечным осложнениям из-за мышечной слабости.
Респиратор
Хроническая дыхательная недостаточность, вторичная по отношению к мышечной слабости, часто приводит к ателектазам, аспирации и пневмонии, вторичной по отношению к ИВЛ.
Сердечный
Кардиомиопатия может быть распространенным сопутствующим заболеванием, и у них может быть хроническая сердечная недостаточность, которая может потребовать медикаментозного лечения.
Опорно-двигательный аппарат
У них может быть сколиоз из-за неподвижности, остеопении и повышенного риска переломов. Также могут развиваться пролежни и контрактуры суставов, даже несмотря на агрессивную физиотерапию.
Эндокринный
У этих пациентов часто встречается надпочечниковая недостаточность, и в случае стресса им может потребоваться прием стероидов. Уровни щитовидной железы и витамина D также следует оценивать в рамках комплексной оценки.
Уровни щитовидной железы и витамина D также следует оценивать в рамках комплексной оценки.
Неврологический
У некоторых из этих пациентов могут быть судороги; Бремя психологических расстройств, таких как депрессия и тревога, у этих пациентов велико из-за основного хронического заболевания.
Ранняя диагностика является ключом к лечению этих расстройств, и при наличии предшествующего семейного анамнеза можно рассмотреть возможность проведения дородового тестирования с анализом ДНК беременной женщины.
Невролог должен осмотреть любого младенца с необъяснимой мышечной слабостью, а генетическое тестирование должно быть частью комплексной оценки, поскольку многие из этих расстройств могут иметь перекрывающиеся признаки.
Установление подтвержденного генетического диагноза необходимо для прогнозирования, консультирования и пренатального тестирования будущих беременностей.
Вскоре после постановки диагноза к уходу за такими пациентами должна быть привлечена межпрофессиональная бригада, поскольку у них могут быть множественные медицинские и психосоциальные проблемы.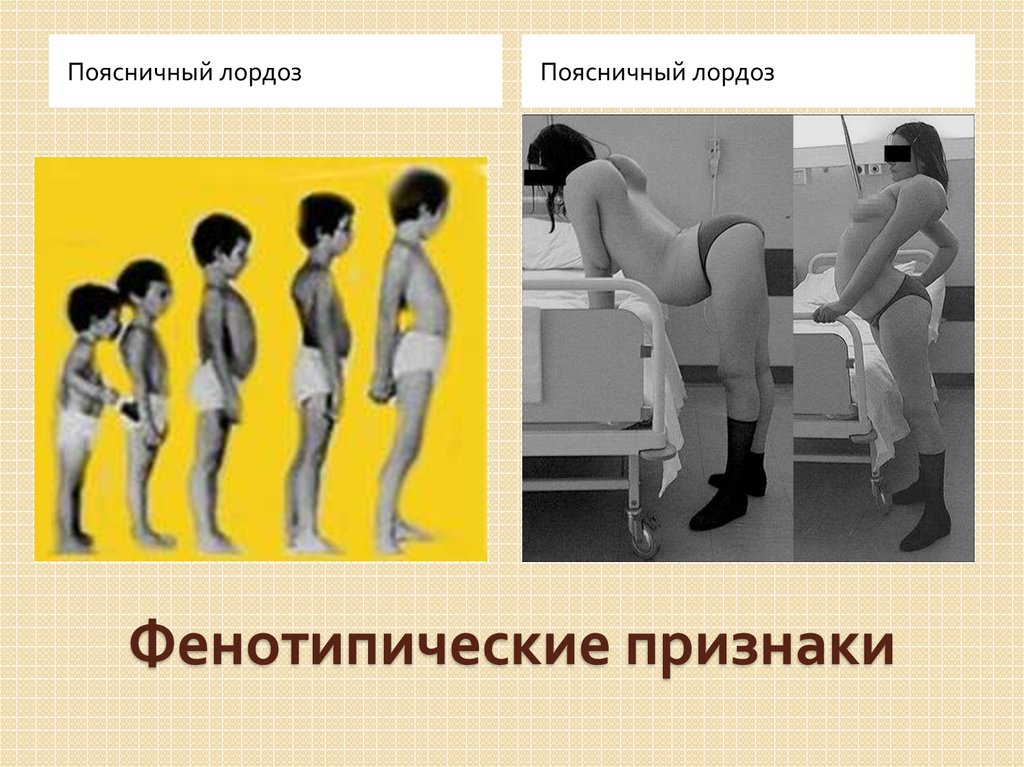 Лица, осуществляющие уход, должны быть хорошо обучены уходу за медицинским оборудованием, которое требуется этим пациентам дома, таким как вентиляторы и трубки для кормления. Для быстрого решения любых основных проблем требуется регулярное последующее наблюдение, и это может улучшить качество жизни этих пациентов.
Лица, осуществляющие уход, должны быть хорошо обучены уходу за медицинским оборудованием, которое требуется этим пациентам дома, таким как вентиляторы и трубки для кормления. Для быстрого решения любых основных проблем требуется регулярное последующее наблюдение, и это может улучшить качество жизни этих пациентов.
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Комментарий к этой статье.
Floriach-Robert M, Cabello A, Simon De Las Heras R, Mateos Beato F. [Неонатальная гипотония мышечного происхождения: анализ 50 случаев]. Неврология. 2001 июнь-июль;16(6):245-53. [PubMed: 11423041]
Ю Э.М., Корнберг А.Дж. Мышечная дистрофия Дюшенна. J Педиатр Здоровье ребенка. 2015 авг; 51 (8): 759-64. [PubMed: 25752877]
Zapata-Aldana E, Ceballos-Sáenz D, Hicks R, Campbell C. Пренатальные, неонатальные и ранние детские особенности врожденной миотонической дистрофии. J нервно-мышечной Dis. 2018;5(3):331-340. [PubMed: 30010141]
J нервно-мышечной Dis. 2018;5(3):331-340. [PubMed: 30010141]
Ши ПБ. Мышечные дистрофии и другие генетические миопатии. Нейрол клин. 2013 ноябрь;31(4):1009-29. [PubMed: 24176421]
Mah JK, Korngut L, Fiest KM, Dykeman J, Day LJ, Pringsheim T, Jette N. Систематический обзор и метаанализ эпидемиологии мышечных дистрофий. Может J Neurol Sci. 2016 Январь; 43 (1): 163-77. [В паблике: 26786644]
Окада М., Кавахара Г., Ногучи С., Суги К., Мураяма К., Нонака И., Хаяши Ю.К., Нишино И. Первичный дефицит коллагена VI является второй наиболее распространенной врожденной мышечной дистрофией в Японии. Неврология. 04 сентября 2007 г .; 69 (10): 1035-42. [PubMed: 17785673]
Schorling DC, Kirschner J, Bönnemann CG. Врожденные мышечные дистрофии и миопатии: обзор и обновление. нейропедиатрия. 2017 авг; 48 (4): 247-261. [PubMed: 28669131]
Кобаяши О. , Хаяши Ю., Арахата К., Одзава Э., Нонака И. Врожденная мышечная дистрофия: клиническое и патологическое исследование 50 пациентов с классической (западной) мерозин-положительной формой. Неврология. 1996 март; 46 (3): 815-8. [PubMed: 8618689]
, Хаяши Ю., Арахата К., Одзава Э., Нонака И. Врожденная мышечная дистрофия: клиническое и патологическое исследование 50 пациентов с классической (западной) мерозин-положительной формой. Неврология. 1996 март; 46 (3): 815-8. [PubMed: 8618689]
Philpot J, Sewry C, Pennock J, Dubowitz V. Клинический фенотип при врожденной мышечной дистрофии: корреляция с экспрессией мерозина в скелетных мышцах. Нервно-мышечное расстройство. 1995 г., июль; 5(4):301-5. [В паблике: 7580243]
Bönnemann CG. Миопатии, связанные с коллагеном VI, врожденная мышечная дистрофия Ульриха и миопатия Бетлема. Handb Clin Neurol. 2011;101:81-96. [Бесплатная статья PMC: PMC5207779] [PubMed: 21496625]
Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé FM, Schwartz K, Fardeau M, Tryggvason K. Мутации в гене 2-цепи ламинина альфа (LAMA2) вызывают врожденную мышечную дистрофию с дефицитом мерозина.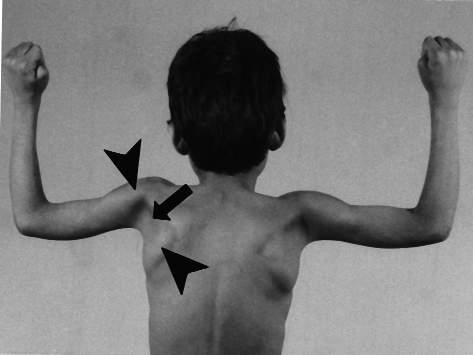 Нат Жене. 1995 октября; 11 (2): 216-8. [PubMed: 7550355]
Нат Жене. 1995 октября; 11 (2): 216-8. [PubMed: 7550355]
Falsaperla R, Praticò AD, Ruggieri M, Parano E, Rizzo R, Corsello G, Vitaliti G, Pavone P. Врожденная мышечная дистрофия: от мышц к мозгу. Ital J Pediatr. 2016 31 августа; 42(1):78. [Бесплатная статья PMC: PMC5006267] [PubMed: 27576556]
Исигаки К., Ихара С., Накамура Х., Мори-Йошимура М., Маруо К., Танигучи-Икеда М., Кимура Э., Мураками Т., Сато Т. , Тода Т., Кайя Х., Осава М. Национальный реестр пациентов с врожденной мышечной дистрофией Фукуямы в Японии. Нервно-мышечное расстройство. 2018 Октябрь; 28 (10): 885-893. [PubMed: 30220444]
Спаркс ЮВ. Неонатальная гипотония. Клин Перинатол. 2015 июнь;42(2):363-71, ix. [PubMed: 26042909]
Барраса-Флорес П., Бейтс Ч.Р., Оливейра-Сантос А., Буркин Д.Дж. Ламинин и интегрин при врожденной мышечной дистрофии, связанной с LAMA2: от болезни к терапии. Фронт Мол Невроски.