







2011-2017 © МБУЗ ГКП № 7, г.Челябинск.
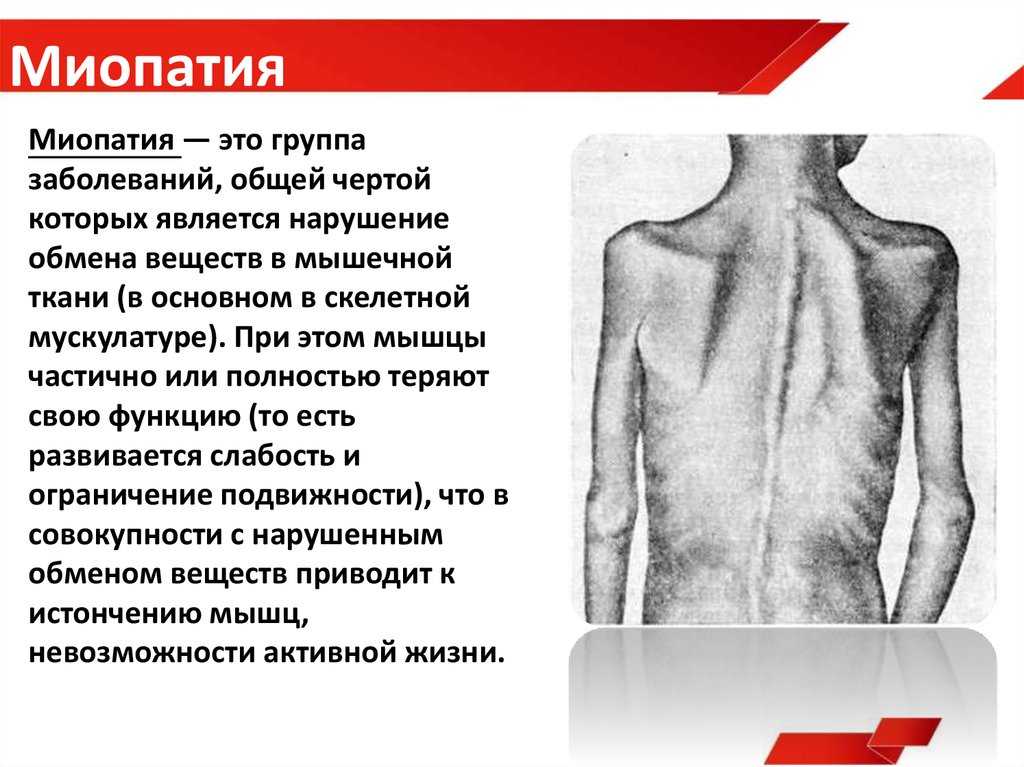
Миодистрофия Дюшенна – наследственное нервно-мышечное заболевание, которое проявляется у мальчиков и характеризуется прогрессирующей мышечной слабостью и утомляемостью, симметричными псевдогипертрофиями голеней и симметричными атрофиями других мышц в сочетании с костно-суставными деформациями, сердечно-сосудистыми и дыхательными нарушениями. Миодистрофия Дюшенна имеет Х-сцепленный рецессивный тип наследования, Английской аббревиатурой DMD обозначается как название гена дистрофина, так и название заболевания – мышечная дистрофия Дюшенна.
Если вы родственник, врач или просто знаете человека с мышечной дистрофией Дюшенна или Беккера, расскажите ему, пожалуйста, про Национальный регистр фонда «МойМио». В Регистре мы объединяем данные о всех пациентах с МДД/МДБ в России. Данные позволят собирать статистику для аргументации проблем редкого пациента, что в свою очередь повысит уровень аргументации в защите прав людей с мышечной дистрофией Дюшенна/Беккера, поможет выстраивать модель оказания медицинской помощи, а значит помогать таким пациентам больше и эффективнее.
Вместе мы сильнее, присоединяйтесь!
Ген дистрофина DMD локализован на коротком плече Х-хромосомы (локус Xp21. 2–р21.1). Это один из самых больших генов человека, поэтому в нем так часто возникают спорадические мутации (до 40%). Заболевание в 60% случаев наследуется мальчиками от женщин-носительниц, как типичное Х-сцепленное рецессивное наследование.
2–р21.1). Это один из самых больших генов человека, поэтому в нем так часто возникают спорадические мутации (до 40%). Заболевание в 60% случаев наследуется мальчиками от женщин-носительниц, как типичное Х-сцепленное рецессивное наследование.
Поэтому мы часто говорим, миодистрофия Дюшенна– болезнь мальчиков. У женщин 2 Х-хромосомы и в случае, если возникнет поломка гена на одной хромосоме, вторая будет вырабатывать дистрофин.
Отсутствие дистрофина (так происходит при мышечной дистрофии Дюшенна) влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Патогенез миодистрофии Дюшенна, включает и иммунопатологические процессы. В организме нарушен процесс регенерации и почти сразу после рождения запускается каскад воспалительной реакции. Из-за возникающего дефекта липидного слоя сарколеммы повышается ее проницаемость, что провоцирует рабдомиолиз. Через отсутствующий защитный барьер клетки внутриклеточная КФК выходит в кровь, а внеклеточный кальций в миоциты.
Из-за возникающего дефекта липидного слоя сарколеммы повышается ее проницаемость, что провоцирует рабдомиолиз. Через отсутствующий защитный барьер клетки внутриклеточная КФК выходит в кровь, а внеклеточный кальций в миоциты.
Выделяют 5 стадий заболевания.
| Стадия | Описание, признаки | |
|---|---|---|
| 1 | Досимптомная | Повышение уровней КФК, ЛДГ, АСТ и АЛТ в биохимическом анализе крови. |
| 2 | Ранняя (амбулаторная) Сохранена способность к самостоятельному передвижению | Использование приёмов Говерса при вставании, миопатическая «утиная» походка, ходьба на носках. Ребёнок поднимается по ступенькам приставным шагом и\или с поддержкой. Ребёнок поднимается по ступенькам приставным шагом и\или с поддержкой. |
| 3 | Поздняя (амбулаторная) Сохранена способность к самостоятельному передвижению | Нарастающие трудности при ходьбе, потеря способности подниматься по ступенькам и вставать с пола. |
| 4 | Ранняя (не амбулаторная) Утрачена способность к самостоятельному передвижению | Пациент способен некоторое время передвигаться самостоятельно, способен удерживать положение тела. Высокие риски развития скелетных деформаций, кардиомиопатии и дыхательных нарушений. |
| 5 | Поздняя (не амбулаторная) Утрачена способность к самостоятельному передвижению | Нарастание ограничения функций верхних конечностей, скелетных деформаций, кардиомиопатии и дыхательных нарушений. Трудности с удержанием положения тела. Трудности с удержанием положения тела. |
Новорожденные с миодистрофией Дюшенна могут не иметь значительных или заметных отклонений. Тогда, первые месяцы и даже год моторное развитие ребенка происходит в пределах нормы или с незначительной задержкой. До 30% пациентов на первом году жизни имеют отставание в психоречевом развитии. Также у таких детей, чаще, чем в среднем в популяции, выявляются расстройства аутистического спектра.
До 10% пациентов с ПМД Дюшенна могут иметь клинические проявления на 1-м году жизни в виде симптомокомплекса «вялого ребенка». Основными симптомами являются слабость мышц тазового пояса, патологическая мышечная утомляемость при физической нагрузке. Изменение походки по типу «утиной» – к 4-5 годам формируется дефект походки: больной широко расставляет ноги, при ходьбе переваливается из стороны в сторону, передвигается на носочках, пациенты помогают себе руками, сильно размахивая ими при ходьбе. В среднем пациенты с миопатией Дюшенна теряют способность самостоятельно передвигаться с 8 до 12 лет. Однако бывают потерявшие способность к самостоятельному передвижению пациенты и в 6 лет, и пациенты, самостоятельно передвигающиеся в 16-17 лет. По всей видимости, это зависит от индивидуальных особенностей течения заболевания.
В среднем пациенты с миопатией Дюшенна теряют способность самостоятельно передвигаться с 8 до 12 лет. Однако бывают потерявшие способность к самостоятельному передвижению пациенты и в 6 лет, и пациенты, самостоятельно передвигающиеся в 16-17 лет. По всей видимости, это зависит от индивидуальных особенностей течения заболевания.
Сердечно-сосудистые расстройства — лабильность пульса, АД, приглушение тонов, расширение границ сердца, сердечная недостаточность. Примерно 73% популяции имеют проявления кардиальной патологии. Сердечно-сосудистая система вовлекается в патологический процесс достаточно рано. Отмечаются изменения миокарда (блокада ножек пучка Гиса и др.).
Первая ступень диагностики при подозрении на миодистрофию Дюшенна· Исследование активности КФК сыворотки крови.
При миопатии Дюшенна происходит распад миоцитов (рабдомиолиз) и высвобождению в кровь КФК и других продуктов цитолиза. Поэтому уровень КФК крови значительно повышен. Отклонения уровня активности КФК в 100 и более раз. Таким образом, исследование активности КФК сыворотки крови может быть первый шагом в диагностике заболевания.
Поэтому уровень КФК крови значительно повышен. Отклонения уровня активности КФК в 100 и более раз. Таким образом, исследование активности КФК сыворотки крови может быть первый шагом в диагностике заболевания.
Кроме определения КФК в биохимическом анализе крови необходимо определять активность таких ферментов как лактатдегидрогеназа (ЛДГ), и, так называемых, «печеночных ферментов» аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ). При ПМД Дюшенна активность фермента ЛДГ бывает повышена в 3-5 раз. Активность ферментов АСТ и АЛТ, имеющих внепеченочное происхождение, может быть повышена в десятки раз.
Для верификации диагноза Прогрессирующая мышечная дистрофия Дюшенна необходима генетическая диагностика.
Проведение ДНК анализаВыполнение генетического исследования начинают обычно с простого метода MLPA (Multiplex ligation-dependent probe amplification – англ. , множественная лиганд-зависимая проба амплификации), позволяющего проверить наличие всех 79 экзонов в гене DMD – так может быть обнаружена делеция экзона/ов.
, множественная лиганд-зависимая проба амплификации), позволяющего проверить наличие всех 79 экзонов в гене DMD – так может быть обнаружена делеция экзона/ов.
Выявление дупликаций проводится посредством модифицированной MLPA. Если мутация так и не была выявлена применяют секвенирование гена DMD, для выявления точечных мутаций. Различные виды генетических исследований позволяют получить более подробную информацию об изменениях структуры (мутациях) ДНК при ПМД Дюшенна. Для проведения исследования необходимо 2 мл венозной крови. Подтверждение диагноза результатами генетического исследования позволяет включить ребенка в клинические исследования, а для его родителей выработать рекомендации по пренатальной диагностике при следующих беременностях. После определения мутации (изменения гена дистрофина), матери ребенка предлагается пройти генетическое исследование для выявления носительства мутации в гене DMD. Такая информация важна для других родственников женского пола со стороны матери (ее сестер, дочерей, теток, двоюродных сестер), поскольку они также могут являться носителями данной мутации.
Возможно проведение пренатальной диагностика ПМД Дюшенна методами молекулярно-генетического исследования (материал для исследования у матери необходимо забирать до 12 недели беременности), а также предимплантационная генодиагностика в случае проведения экстракорпорального оплодотворения.
Применяемое в настоящее время противовоспалительное лечение кортикостероидами является золотым стандартом, описанным в международном руководстве по ведению пациентов с миодистрофией Дюшенна.
Данные рандомизированных контролируемых исследований показали пользу кортикостероидов для восстановления мышечной силы. Кроме основного противовоспалительного эффекта (снимает отек и воспаление, стабилизирует мышечную мембрану), терапия кортикостероидами позволяет пациенту продлить время способности к самостоятельному передвижению (ходьбы). В настоящее время прием кортикостероидов сохраняется даже после потери пациента способности ходить. Продолжение терапии поможет сдерживать развитие сколиоза, сохранять мышечную силу в верхних конечностях, поддержать дыхательную и сердечную функцию. Поскольку кортикостероиды используются пациентами с миодистрофией Дюшенна в течение длительного времени, важен мониторинг нежелательных явлений и осложнений такой терапии (например, набор веса, изменения в поведении, задержку роста и\или полового созревания и др.).
В настоящее время прием кортикостероидов сохраняется даже после потери пациента способности ходить. Продолжение терапии поможет сдерживать развитие сколиоза, сохранять мышечную силу в верхних конечностях, поддержать дыхательную и сердечную функцию. Поскольку кортикостероиды используются пациентами с миодистрофией Дюшенна в течение длительного времени, важен мониторинг нежелательных явлений и осложнений такой терапии (например, набор веса, изменения в поведении, задержку роста и\или полового созревания и др.).
Кардиомиопатия и сердечная недостаточность являются основными причинами смертности у пациентов с МДД.
Целью кардиопротективной терапии является снижение нагрузки на миокард. Доказана эффективность назначения ингибиторов АПФ для профилактики дилатационной кардиомиопатии. Например, одним из первых было исследование Института миологии в Париже показавшее, что при раннем назначении ингибиторов АПФ к 15 годам кардиомиопатия сформировалась всего у 20–30 процентов пациентов вместо 70 процентов, как было раньше.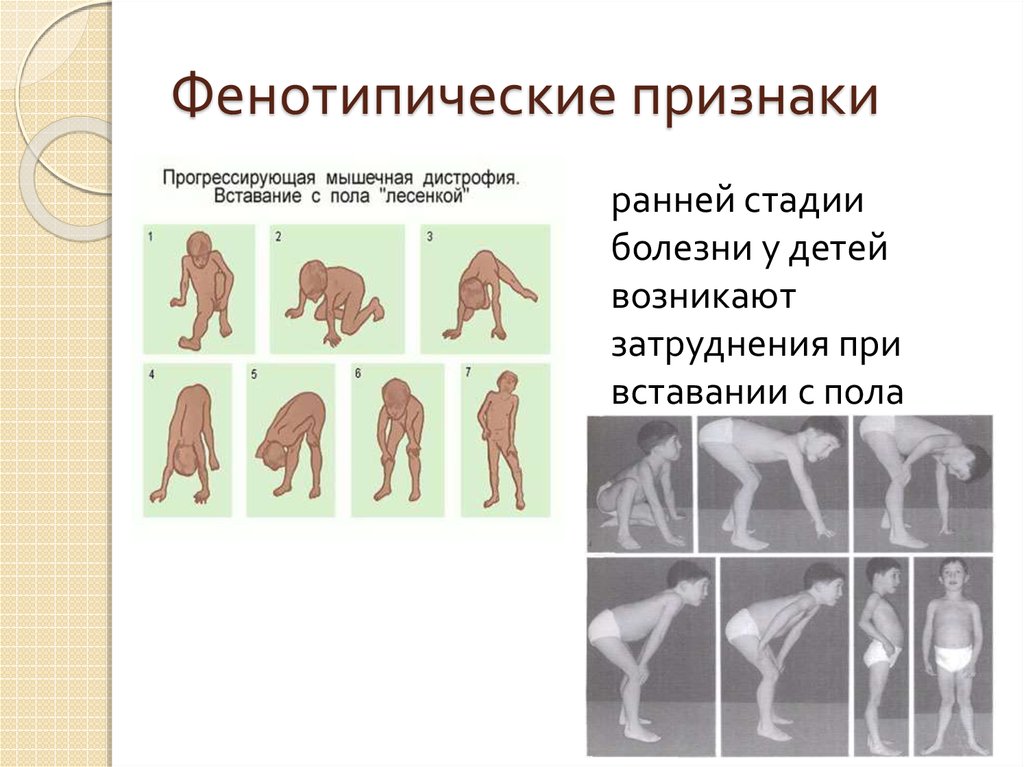 Также назначаются препараты, снижающие частоту сердечных сокращений – бета-блокаторы. Это относится к стадии компенсации.
Также назначаются препараты, снижающие частоту сердечных сокращений – бета-блокаторы. Это относится к стадии компенсации.
На стадии декомпенсации специалист может рекомендовать дополнительные препараты (например, кардиотоники).
Метаболическая терапияДля профилактики остеопороза показано назначение препаратов, содержащих витамин D3 и кальций.
Генная терапияЦель этиотропного лечения устранить причину заболевания — мутацию в гене DMD, тем самым восстановить синтез дистрофина. В настоящее время в мире одобрено несколько препаратов, восстанавливающих синтез белка.
Нонсенс мутация. Стоп-кодонПриблизительно 10–15% МДД вызваны точечными мутациями, приводящими к образованию преждевременного стоп-кодона, то есть, преждевременной остановке синтеза белка дистрофина. Аталурен (Translarna, PTC Therapeutics, США) разработан для пропуска преждевременных стоп-кодонов. Аталурен связывается с рибосомной РНК и ухудшает распознавание преждевременного стоп-кодона, что позволяет восстановить трансляцию и синтез модифицированного дистрофина.
Аталурен (Translarna, PTC Therapeutics, США) разработан для пропуска преждевременных стоп-кодонов. Аталурен связывается с рибосомной РНК и ухудшает распознавание преждевременного стоп-кодона, что позволяет восстановить трансляцию и синтез модифицированного дистрофина.
Нарушение рамки считывания (процесса синтеза белка дистрофин) в большинстве случаев связано с делециями и дупликациями, поэтому основной разрабатываемой стратегией генотерапии являлся метод пропуска экзона или экзонов. Такой метод приводит к восстановлению рамки считывания, иными словами – возобновлению экспрессии укороченного дистрофина. Восстановление экспрессии частично функционального дистрофина должно преобразовать злокачественную форму МДД с тяжелым фенотипическим проявлением в МДБ – мышечную дистрофию Беккера. Данный метод лечения может помочь большинству пациентов с мышечной дистрофией Дюшенна. Исключение составят делеции, разрушающие актин-связывающие домены в N-концевой области или затрагивающие первый или последний экзоны и крупные хромосомные перестройки. Такие мутации достаточно редки и вместе составляют менее 10% от всех описанных мутаций в гене DMD. В настоящее время одобрены три препарата, использующие метод пропуска экзона с помощью антисмысловых олигонуклеотидов АОН.
Такие мутации достаточно редки и вместе составляют менее 10% от всех описанных мутаций в гене DMD. В настоящее время одобрены три препарата, использующие метод пропуска экзона с помощью антисмысловых олигонуклеотидов АОН.
Самым ожидаемым является универсальный препарат или метод, который сможет применяться при любом виде мутации в гене дистрофин.
Таким препаратом можно назвать мини и микродистрофин или метод CRISPR/Cas9.
Внедрение мультидисциплинарного подхода и соблюдение стандартов оказания медицинской помощи позволило увеличить продолжительность жизни пациентов с мышечной дистрофией Дюшенна в мире по данным ретроспективного анализа в среднем до 27,9 лет (от 23 до 38,6 лет).
Ссылки на использованные статьи:Saito T, Kawai M, Kimura E et al. Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul Disord. 2017 Feb;27(2):107-114. doi: 10.1016/j.nmd.2016.11.012.
Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul Disord. 2017 Feb;27(2):107-114. doi: 10.1016/j.nmd.2016.11.012.
Passamano L., Taglia A., Palladino A. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012 Oct; 31(2): 121–125.
Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard JP, Roy R, et al. Dystrophin expression in muscles of Duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65(4):371–86. https://doi.org/10.1097/01. jnen.0000218443.45782
Статья носит информационный, ознакомительный характер. Диагностика заболевания и лечение возможны только под наблюдением специалистов
Изображение от Freepik
Мышечная дистрофия Дюшенна или миопатия Дюшенна — редкое наследственное заболевание, которое передается от матери к сыну. Сами женщины при этом не страдают этой патологией. Болезнь диагностируется у одного из 3500–4000 мальчиков и проявляется в раннем возрасте.
Сами женщины при этом не страдают этой патологией. Болезнь диагностируется у одного из 3500–4000 мальчиков и проявляется в раннем возрасте.
Первопричина мышечной дистрофии — мутация (дупликация или делеция) в гене дистрофина, который расположен в X-хромосоме. Поскольку у мальчиков только одна Х-хромосома, они более подвержены этому заболеванию. У девочек патология встречается, но крайне редко, и выражена она легкими симптомами: поражением миокарда, небольшой слабостью мышц.
Среди женщин проводится анализ на выявление генетической мутации, которая приводит к развитию заболевания у ребенка. Исследование эффективно в 80 % случаев.
Однако нормальный результат анализа не всегда исключает риск развития плода с этим заболеванием:

В России данный анализ делается бесплатно.
Сыновья женщины-носителя гена заболевания имеют 50 % вероятность развития миопатии. Дочери станут носителями того же гена.
Дистрофин — белок, содержащийся в мышечной ткани, — необходим для нормального функционирования мышц. У больных миопатией Дюшенна этого вещества недостаточно. Со временем мышечные волокна повреждаются, ослабляются.
Первые проявления заболевания наблюдаются у детей в возрасте 2–3 лет. Все начинается с поражения нижних конечностей, что сказывается на походке: ребенок ходит вразвалку, начинает передвигаться на пальцах. Детям с этим заболеванием сложно совершать активные действия: бегать, прыгать, вставать с пола и подниматься по лестнице. Они больше подвержены переломам, поскольку чаще остальных падают. Затем патология затрагивает тазовые и спинные мышцы, грудную клетку и верхние конечности. Почти у всех пациентов наблюдается сколиоз и контрактура конечностей.
Почти у всех пациентов наблюдается сколиоз и контрактура конечностей.
При миопатии Дюшенна наблюдается замена отдельных мышечных групп фиброзной или жировой тканью, в частности на лодыжках. К 12 годам для передвижений требуется инвалидная коляска.
К 14 годам у 30 % пациентов и у всех больных мышечной дистрофией старше 18 лет наблюдаются осложнения на сердечную мышцу: аритмия, кардиомиопатия, нарушение проводимости. Однако эти патологии протекают бессимптомно, поскольку больные не способны на физическую активность. Около трети пациентов страдает слабоумием, которое больше сказывается на вербальных способностях, чем на производительности.
Опасность заболевания кроется в поздней диагностике. Большинство родителей замечают миопатию Дюшенна в позднем возрасте, так как не способны сразу определить затруднения передвижения.
Избыточным весом страдают, в основном, дети, которым назначен прием кортикостероидов. У остальных пациентов подросткового и взрослого возраста, напротив, наблюдается дефицит мышечной массы. Поэтому помимо основного лечения больным необходима консультация диетолога. Важно употребление клетчатки для профилактики запоров.
Поэтому помимо основного лечения больным необходима консультация диетолога. Важно употребление клетчатки для профилактики запоров.
На поздних стадиях у многих пациентов появляются проблемы с пережевыванием и глотанием пищи, в тяжелых случаях может потребоваться гастростомия – хирургическое введение трубки в желудок для осуществления питания.
Для диагностики заболевания необходим анализ на наличие мутировавшего гена, в некоторых случаях также назначают мышечную биопсию, электромиографию. Предпосылками к обследованию на миопию Дюшенна становится семейный анамнез, наличие клинических признаков.
При наличии заболевания биопсия показывает изменение размера и некроз мышечных волокон, не отделенных от моторных единиц. Также у больного повышен уровень креатинкиназы (в 100 раз превышает нормальный показатель).
Основным клиническим тестом, подтверждающим заболевание, является исследование мутаций ДНК в лейкоцитах периферической крови.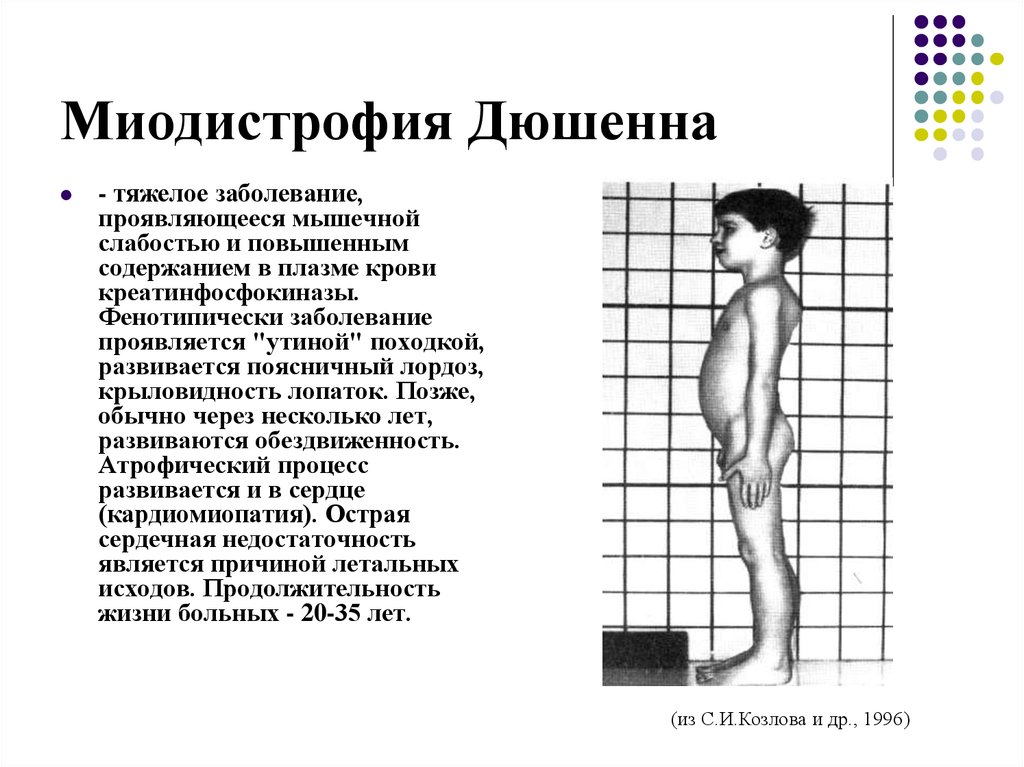 Около 70 % пациентов с мышечной дистрофией имеют делеции дистрофина, а около 10 % — дупликации.
Около 70 % пациентов с мышечной дистрофией имеют делеции дистрофина, а около 10 % — дупликации.
Также для диагностики используется анализ мочи, общий и биохимический анализ крови. Рекомендуется консультация генетика.
Специального лечения миопатии Дюшенна не существует. Больному назначают поддерживающие меры, в некоторых случаях прием дефлазакорта или преднизона, бета-блокаторы, ингибиторы АПФ. Может быть выбран радикальный метод лечения — корректирующая операция (в частности при сколиозе).
Пациентам показаны легкие и пассивные упражнения, которые позволяют дольше сохранить способность к передвижению и предупреждают развитие дисфункциональной атрофии. Для предотвращения контрактур показаны ортопедические вмешательства. Например, ношение ортеза голеностопного сустава во время сна позволяет предупредить сгибательные контрактуры.
Пациентам с миопатией Дюшенна важно поддерживать гармоничное питание, чтобы избежать ожирения: из-за снижения двигательных функций у людей с этим заболеванием ниже потребность в калориях.
Иногда для лечения дыхательной недостаточности используют назальные маски и другие виды неинвазивной респираторной поддержки. Все большее распространение получает элективная трахеотомия (хирургический разрез трахеи для создания альтернативного пути дыхания). Эта процедура позволяет больным миопатией Дюшенна продлить жизнь.
Пациентам старше 5 лет, двигательные навыки которых остановились в развитии или регрессируют, назначают ежедневный прием дефлазакорта или преднизона. Спустя 10 дней после начала курса препараты начинают действовать. Пик эффективности приходится на 3 месяца с начала терапии и сохраняется в течение следующих 6 месяцев.
При долгосрочном применении у пациента повышается выносливость, двигательные способности он утрачивает на 1,4–2,5 года позже. Также лечение позитивно сказывается на работе легких, снижает необходимость в хирургической коррекции сколиоза и других ортопедических осложнений, стабилизирует работу сердечной мышцы, повышает выживаемость.
Среди побочных эффектов приема преднизона – набор массы (проявляется в интервале 6–18 месяцев с момента начала приема). Также увеличивается риск перелома длинных костей, позвоночника.
Миопатия Дюшенна – неизлечимое заболевание с неблагоприятным прогнозом.
Ранее средняя продолжительность жизни пациентов с мышечной дистрофией составляла не более 23 лет. На сегодняшний день пациенты с этим диагнозом живут 27 лет и более при условии постоянной и грамотной терапии.
Продолжительность жизни также зависит от наличия сопутствующих заболеваний. Постепенно мышечная слабость провоцирует развитие сердечных патологий и проблем с дыханием. Самые распространенные осложнения, которые ухудшают прогноз и сокращают жизнь пациента, — заболевания дыхательных путей.
Перейти к основному содержанию
Слабость, связанная с мышечной дистрофией Дюшенна (МДД), избирательно поражает мышцы конечностей, расположенные ближе к туловищу, прежде чем мышцы, расположенные дальше от него; ноги поражаются раньше рук.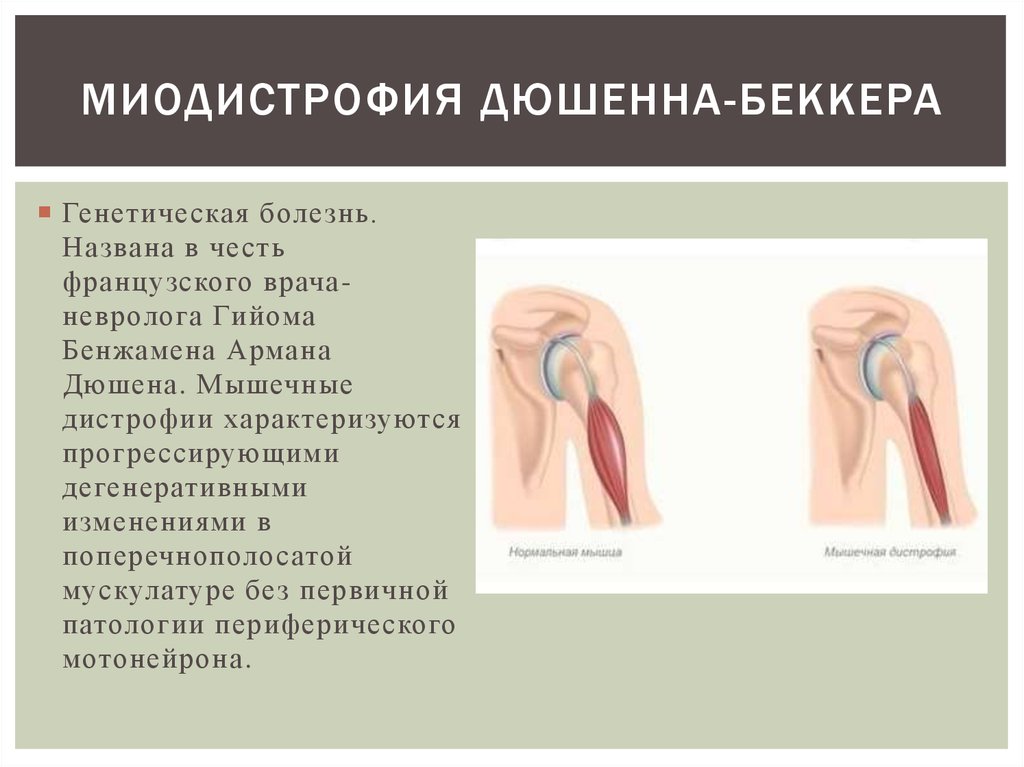 Скорость роста при МДД обычно ниже нормы в первые годы жизни, что приводит к низкорослости.
Скорость роста при МДД обычно ниже нормы в первые годы жизни, что приводит к низкорослости.
Мальчики с МДД часто поздно ходят.
Известно, что вариации гена LTBP4 и регуляторной области гена SPP1 влияют на возраст потери способности передвигаться и/или снижение мышечной силы. 1,2,3,4,5 У малышей родители могут заметить увеличение икроножных мышц (см. изображение справа). Это увеличение известно как псевдогипертрофия или «ложное увеличение», поскольку мышечная ткань является аномальной. Псевдогипертрофия также может возникать в мышцах бедер. Псевдогипертрофия может возникать и в мышцах бедер.
Дошкольник с МДД может казаться неуклюжим и часто падать. Родители также могут заметить, что детям трудно подниматься по лестнице, вставать с пола или бегать. Вставая с пола, больные мальчики могут использовать поддержку рук, чтобы подняться в вертикальное положение.
К школьному возрасту дети могут ходить на носочках или подушечках стоп, слегка переваливаясь, и часто падать. Чтобы сохранить равновесие, они могут выпятить живот и отвести плечи назад. Дети также испытывают трудности с поднятием рук.
Чтобы сохранить равновесие, они могут выпятить живот и отвести плечи назад. Дети также испытывают трудности с поднятием рук.
Многие дети с МДД начинают пользоваться инвалидной коляской где-то к 12 годам. Переход на инвалидную коляску обычно является постепенным процессом; на первых порах кресло может понадобиться только для экономии сил ребенка при преодолении дальних расстояний. Дети часто обретают новую независимость после того, как полностью пересаживаются на инвалидную коляску с электроприводом.
В подростковом возрасте при выполнении движений руками, ногами или туловищем может потребоваться помощь или механическая поддержка.
Пациенты с МДД часто умирают в подростковом возрасте или в возрасте 20 лет от дыхательной недостаточности или кардиомиопатии; только несколько пациентов с МДД выживают после третьего десятилетия жизни.
Поражение мышц при МДД само по себе обычно не вызывает боли. Некоторые люди время от времени жалуются на мышечные спазмы; их обычно можно лечить безрецептурными болеутоляющими средствами.
Поскольку мышечная дистрофия не влияет непосредственно на нервы, осязание и другие чувства остаются нормальными, равно как и контроль над гладкими, или непроизвольными, мышцами мочевого пузыря и кишечника, а также половые функции.
Недостаток дистрофина может ослабить мышечный слой сердца ( myocardium ), что приводит к состоянию, называемому кардиомиопатией , характеризующемуся обширным рубцеванием ткани. МДД также может вызывать нарушения проводимости в сердце. Сообщалось, что все пациенты старше 18 лет имеют симптомы кардиомиопатии. Со временем, иногда уже в подростковом возрасте, ущерб, наносимый МДД сердцу, может стать опасным для жизни. Сердце должно тщательно контролироваться, как правило, детским кардиологом. Дополнительную информацию о кардиомиопатии при МДД см. в разделе Медицинское управление.
Серийный мониторинг дыхательной способности следует начинать в возрасте 5 или 6 лет. Диафрагма и другие мышцы, управляющие легкими, могут ослабевать, что делает легкие менее эффективными при продвижении воздуха внутрь и наружу. Хотя ребенок может не жаловаться на одышку, проблемы, которые указывают на плохую дыхательную функцию, включают головные боли, умственную тупость, трудности с концентрацией внимания или бодрствованием, а также ночные кошмары. Дети, прикованные к инвалидной коляске, как правило, имеют признаки плохой функции легких.
Хотя ребенок может не жаловаться на одышку, проблемы, которые указывают на плохую дыхательную функцию, включают головные боли, умственную тупость, трудности с концентрацией внимания или бодрствованием, а также ночные кошмары. Дети, прикованные к инвалидной коляске, как правило, имеют признаки плохой функции легких.
Ослабленные дыхательные мышцы затрудняют кашель, что приводит к повышенному риску серьезной респираторной инфекции. Простая простуда может быстро перейти в пневмонию. Важно сделать прививку от гриппа, а в случае заражения – незамедлительно начать лечение. См. «Медицинское управление» для получения дополнительной информации о респираторной помощи при МДД.
Около трети мальчиков с МДД имеют некоторую степень нарушения обучаемости , хотя лишь немногие имеют серьезные когнитивные нарушения. Врачи считают, что аномалии дистрофина в головном мозге могут оказывать незначительное влияние на когнитивные функции и поведение. Проблемы с обучением при МДД возникают в трех основных областях: концентрация внимания, вербальное обучение и память, а также эмоциональное взаимодействие.
Дети с подозрением на нарушение способности к обучению могут быть обследованы нейропсихологом, занимающимся развитием, или детским нейропсихологом в отделе специального образования школьной системы или по направлению от MDA Care Center.
Если диагностирована неспособность к обучению, образовательные и психологические вмешательства могут быть начаты немедленно. Специалист может прописать упражнения и техники, которые помогут улучшить эти области, а школы также могут оказать специальную помощь в обучении. Дополнительные сведения о нарушениях обучаемости при МДД см. в разделе Медицинское управление.
 Дж. Нейрол. Нейрохирург. Психиатрия (2015). doi:10.1136/jnnp-2014-308409
Дж. Нейрол. Нейрохирург. Психиатрия (2015). doi:10.1136/jnnp-2014-308409 Штат или почтовый индекс
Español
DMD; Мышечная дистрофия, Дюшенна; Мышечная дистрофия, псевдогипертрофическая прогрессирующая, МДД типа Дюшенна; Мышечная дистрофия, Дюшенна; Muscular dystrophy, pseudohypertrophic progressive, Duchenne type
Мышечная дистрофия Дюшенна (МДД) поражает мышцы, приводя к их истощению, которое со временем ухудшается. МДД встречается в основном у мужчин, хотя в редких случаях может поражать женщин. Симптомы МДД включают прогрессирующую слабость и потерю (атрофию) как скелетной, так и сердечной мышцы. Ранние признаки могут включать задержку способности сидеть, стоять или ходить, а также трудности с обучением речи. Мышечная слабость обычно заметна в раннем детстве. МДД вызывается генетическими изменениями (вариантами ДНК) в гене МДД. МДД наследуется по рецессивному типу, сцепленному с Х-хромосомой, и может возникать у людей, не имеющих семейной истории МДД. Диагноз МДД ставится на основании симптомов, клинического осмотра и результатов биопсии с целью удаления небольшого кусочка мышцы для исследования под микроскопом. Результат генетического тестирования также может помочь подтвердить диагноз. Мышечная дистрофия Беккера (МДБ), более легкая форма мышечной дистрофии, также вызывается генетическими изменениями в гене МДД.
МДД встречается в основном у мужчин, хотя в редких случаях может поражать женщин. Симптомы МДД включают прогрессирующую слабость и потерю (атрофию) как скелетной, так и сердечной мышцы. Ранние признаки могут включать задержку способности сидеть, стоять или ходить, а также трудности с обучением речи. Мышечная слабость обычно заметна в раннем детстве. МДД вызывается генетическими изменениями (вариантами ДНК) в гене МДД. МДД наследуется по рецессивному типу, сцепленному с Х-хромосомой, и может возникать у людей, не имеющих семейной истории МДД. Диагноз МДД ставится на основании симптомов, клинического осмотра и результатов биопсии с целью удаления небольшого кусочка мышцы для исследования под микроскопом. Результат генетического тестирования также может помочь подтвердить диагноз. Мышечная дистрофия Беккера (МДБ), более легкая форма мышечной дистрофии, также вызывается генетическими изменениями в гене МДД.
По оценкам, в США это заболевание составляет менее
Информация о многих редких заболеваниях ограничена. В настоящее время GARD может предоставить следующую информацию по этому заболеванию:
* В настоящее время данные могут быть недоступны для GARD.
Неврологическое заболевание
Возраст, в котором чаще всего появляются симптомы болезни, называется возрастом начала заболевания. Возраст начала может варьироваться для разных заболеваний и может использоваться врачом для определения диагноза. При некоторых заболеваниях симптомы могут проявляться в одном или нескольких возрастных диапазонах.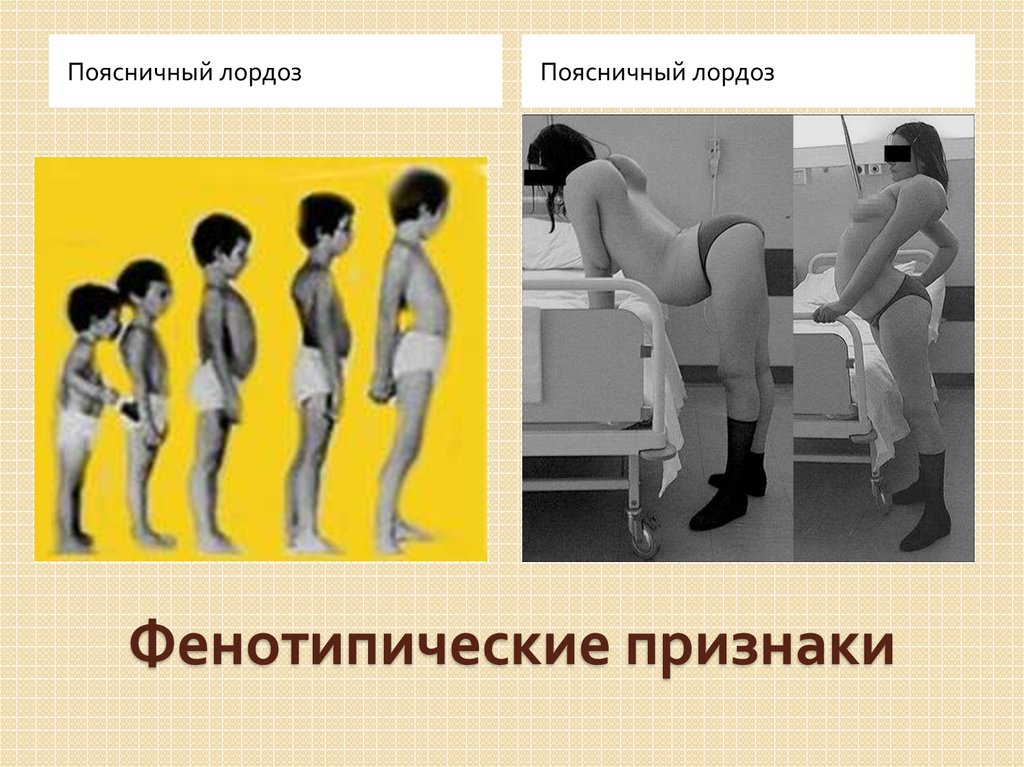 При других заболеваниях симптомы могут появиться в любой момент жизни человека.
При других заболеваниях симптомы могут появиться в любой момент жизни человека.
Пренатальный период
До рождения
Новорожденный
От рождения до 4 недель
Младенец
От 1 до 23 месяцев
Ребенок Выбрано
2-11 лет
Подростки
12-18 лет
Взрослые
19-65 лет
Пожилые Взрослые цветные значки.
14 Симптомы
Просмотр плитки
Просмотр списка
Просмотр плитки
Просмотр списка
Незнакомил
Очень часто
.0002 Всегда
Мышечная гипертрофия, поражающая икроножные мышцы.
Мышечная гипертрофия, поражающая икроножные мышцы.
Мышечная гипертрофия икроножных мышц
Мышечная гипертрофия икроножных мышц
14 Симптомы
Эта информация взята из Онтологии фенотипов человека (HPO) что это вызвано тем, что один или несколько генов не работают должным образом.
Disease causing variants in the following gene(s) are known to cause this disease: DMD
Questions:
Answer
What is a gene?
Гены являются частью нашей ДНК, основного генетического материала, содержащегося в каждой клетке нашего тела. Клетки являются строительными блоками всех живых существ, а специализированные клетки образуют органы и ткани нашего тела. ДНК находится в ядре клетки и у человека упакована в 23 пары хромосом с помощью специальных белков.
Каждый ген выполняет свою работу в наших клетках. Некоторые гены служат инструкциями для производства белков. Белки необходимы для структуры, функционирования и регуляции клеток, тканей и органов организма. Некоторые гены могут включать или выключать другие гены. Другие производят молекулы РНК, которые участвуют в химических реакциях в организме.
Некоторые гены могут включать или выключать другие гены. Другие производят молекулы РНК, которые участвуют в химических реакциях в организме.
Источники для получения дополнительной информации: что такое ген? (МедлайнПлюс); Что такое ген? (НГРИ) ; Что такое белки и что они делают? (MedlinePlus)Гены являются частью нашей ДНК, основного генетического материала, содержащегося в каждой клетке нашего тела. Клетки являются строительными блоками всех живых существ, а специализированные клетки образуют органы и ткани нашего тела. ДНК находится в ядре клетки и у человека упакована в 23 пары хромосом с помощью специальных белков.
Каждый ген выполняет свою работу в наших клетках. Некоторые гены служат инструкциями для производства белков. Белки необходимы для структуры, функционирования и регуляции клеток, тканей и органов организма. Некоторые гены могут включать или выключать другие гены. Другие производят молекулы РНК, которые участвуют в химических реакциях в организме.
Источники для получения дополнительной информации: что такое ген? (МедлайнПлюс); Что такое ген? (НГРИ) ; Что такое белки и что они делают? (MedlinePlus)
Подробнее
Все люди наследуют две копии большинства генов.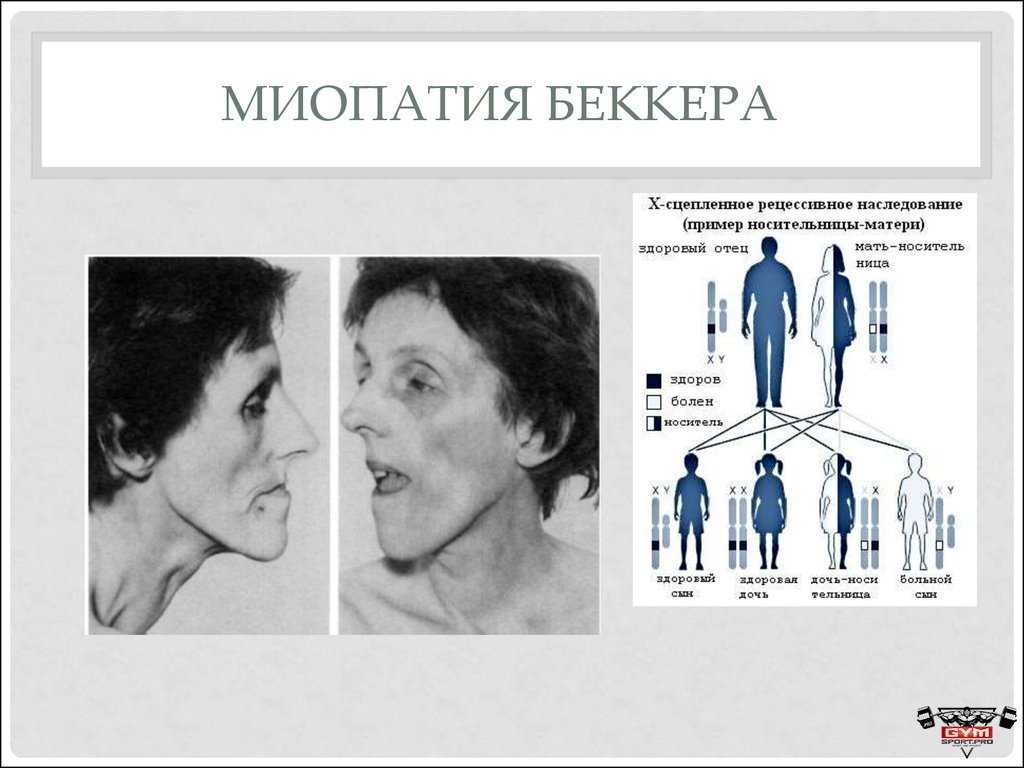 Количество копий гена, которые должны иметь болезнетворный вариант, влияет на способ наследования болезни. Это заболевание наследуется по следующей схеме:
Количество копий гена, которые должны иметь болезнетворный вариант, влияет на способ наследования болезни. Это заболевание наследуется по следующей схеме:
Вопросы:
Описание
Х-сцепленное рецессивное наследование половые хромосомы. Гены, как и хромосомы, обычно идут парами. Рецессивный означает, что при наличии двух копий ответственного гена обе копии должны иметь болезнетворное изменение (патогенный вариант), чтобы у человека было заболевание. Мутация — это более старый термин, который до сих пор иногда используется для обозначения патогенного варианта.
Поскольку женщины имеют две Х-хромосомы, патогенный вариант Х-линейного рецессивного заболевания обычно должен присутствовать в обеих копиях гена, чтобы вызвать заболевание. Поскольку у мужчин есть одна Х-хромосома и, следовательно, только одна копия гена, патогенного варианта в их одной копии достаточно, чтобы вызвать заболевание. Женщин, имеющих патогенный вариант в одной копии гена, называют носителями. В редких случаях у женщин-носителей могут наблюдаться симптомы от легких до умеренных, но у большинства из них симптомы отсутствуют.
Женщин, имеющих патогенный вариант в одной копии гена, называют носителями. В редких случаях у женщин-носителей могут наблюдаться симптомы от легких до умеренных, но у большинства из них симптомы отсутствуют.
Женщина-носитель одного варианта гена, сцепленного с Х-хромосомой, имеет 50% (1 из 2) шансов родить сына с заболеванием и 50% (1 из 2) шансов родить дочь-носителя. Мужчина с Х-сцепленным рецессивным заболеванием не может передать заболевание своим сыновьям, но все его дочери будут носителями. Если ребенок мужского пола является первым человеком в семье с заболеванием, то патогенный вариант мог быть унаследован от матери или случайно впервые возник у ребенка (de novo). X-сцепленный означает, что ген расположен на Х-хромосоме, одной из двух половых хромосом. Гены, как и хромосомы, обычно идут парами. Рецессивный означает, что при наличии двух копий ответственного гена обе копии должны иметь болезнетворное изменение (патогенный вариант), чтобы у человека было заболевание. Мутация — это более старый термин, который до сих пор иногда используется для обозначения патогенного варианта.
Поскольку женщины имеют две Х-хромосомы, патогенный вариант Х-линейного рецессивного заболевания обычно должен присутствовать в обеих копиях гена, чтобы вызвать заболевание. Поскольку у мужчин есть одна Х-хромосома и, следовательно, только одна копия гена, патогенного варианта в их одной копии достаточно, чтобы вызвать заболевание. Женщин, имеющих патогенный вариант в одной копии гена, называют носителями. В редких случаях у женщин-носителей могут наблюдаться симптомы от легких до умеренных, но у большинства из них симптомы отсутствуют.
Женщина-носитель одного варианта гена, сцепленного с Х-хромосомой, имеет 50% (1 из 2) шансов родить сына с заболеванием и 50% (1 из 2) шансов родить дочь-носителя. Мужчина с Х-сцепленным рецессивным заболеванием не может передать заболевание своим сыновьям, но все его дочери будут носителями. Если ребенок мужского пола является первым человеком в семье с заболеванием, патогенный вариант мог быть унаследован от матери или случайно возник у ребенка впервые (de novo).