



2011-2017 © МБУЗ ГКП № 7, г.Челябинск.
Автор статьи
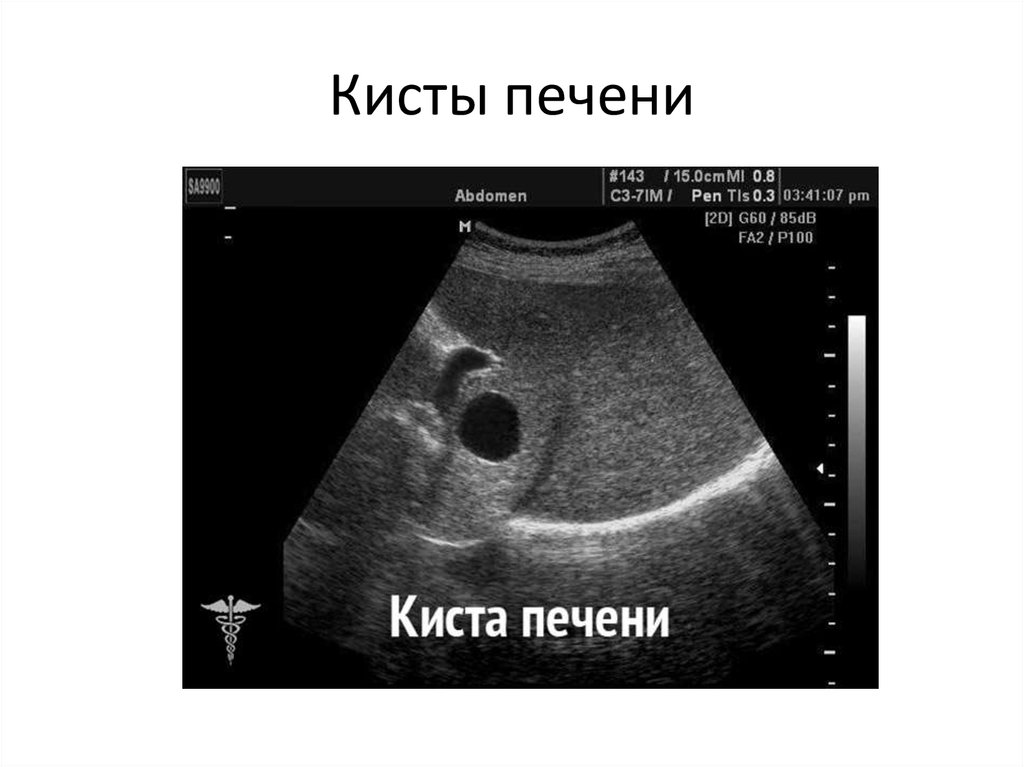
Киста печени – полость в печени разных размеров, которая заполнена жидкостью. Как правило, является доброкачественным образованием, протекает бессимптомно, обнаруживается случайно во время обследования. Киста больших размеров, может нарушать работу печени и окружающих органов.
Распространенность кист невысока, около 5 % населения подвержены развитию патологии.
Неинфекционные кисты чаще встречаются среди женского населения, имеют тенденцию формироваться с возрастом. Эхинококковые (инфекционные) кисты печени развиваются в эндемичных для эхинококка регионах: Россия и страны СНГ, восточное Средиземноморье, Южная Америка, западный Китай.
Кисты, которые сопровождаются симптомами, лечат хирургическим путем, в остальных случаях требуется динамическое наблюдение.
Важным в постановке диагноза и определении тактики лечения является междисциплинарный подход. Радиологи, гастроэнтерологи, хирурги, инфекционисты, — командная, координированная работа важна для эффективности лечения.
Киста печени, которая стремительно увеличивается в размерах и количестве, может вызывать следующие симптомы:
Природа формирования простых однокамерных кист до конца остается неясной. Формирование кистозных полостей может быть результатом нарушения внутриутробного развития желчных протоков.
Некоторые виды кист связаны с серьезной патологией:
Полостные образования классифицируют следующим образом:
Как указано выше, зачастую кистозные образования обнаруживаются случайно.
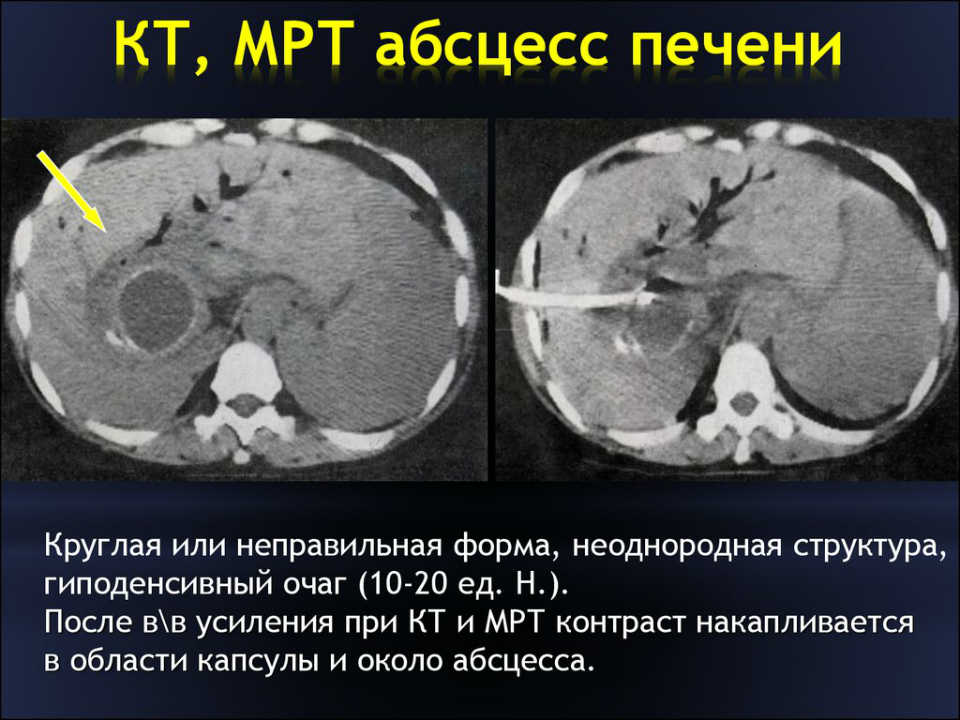
Наравне с ультразвуковым исследованием на практике применяются компьютерная томография и магнитно-резонансная томография.
Диагноз ставится на основании опроса, выявления жалоб, а также тщательного осмотра. Врач может задать вопросы о наличии хронических заболеваний, перенесенных травмах, операциях, проживании в эндемичных по эхинококкозу регионах страны.
При достижении больших размеров образование может быть обнаружено при пальпации врачом передней брюшной стенки.
При необходимости выполняют лабораторную диагностику (определение специфических антител к антигену эхинококковой инфекции в крови), УЗИ с контрастированием для усиления визуализации структур, кровотока, а также пункцию с последующим цитологическим исследованием.
Важно отличать кисту печени от других заболеваний желудочно-кишечного тракта, которые дают схожую симптоматику: желчнокаменная болезнь, гастроэзофагеальный рефлюкс, пептическая язва, гемангиома или гамартома печени.
Тактика зависит от наличия симптомов, размеров, количества, а также сопутствующей патологии.
Бессимптомные кисты, которые достигают 4 см., не нуждаются в лечении, требуют наблюдения в динамике. УЗИ проводят с 3-12 месячным интервалом. При стабильном состоянии в течение трёх лет динамическое наблюдение прекращается.
Органосохраняющие операции предпочтительны:
 Иногда крупные кисты могут рецидивировать и требуют повторного дренирования
Иногда крупные кисты могут рецидивировать и требуют повторного дренированияПолное иссечение или гепатэктомия – радикальные методы, должны выполняться в случае острой необходимости.
Несмотря на то, что данная патология характеризуется медленным ростом и прогрессированием, к ней нужно относиться с максимальным внимание и осторожностью. При достижении больших размеров киста может разорваться. В этом случае может присоединиться бактериальный возбудитель с развитием тяжелой инфекции.
Эхинококкоз имеет специфические осложнения, главное из которых анафилактический шок с потенциальным летальным исходом.
Большие кисты могут нагнаиваться, увеличивая риск развития кровотечения и нарушение оттока желчи по протокам. В литературе описаны редкие случаи озлокачествления.
Прогноз зависит от характера кистозных изменений. За бессимптомными кистами нужно наблюдать в динамике с частотой, установленной лечащим врачом. После полноценного хирургического лечения рецидивы отмечаются редко. Профилактика кист печени заключается в регулярном диспансерном наблюдении. При возникновении подозрительных симптомов необходимо обратиться за медицинской помощью.
Внимание в вопросе профилактики стоит уделить эхинококкозу. Эхинококкоз – паразитарная инфекционная патология, заражение человека происходит после контакта с зараженными животными (собаки, коровы, лошади, свиньи), употреблении в пищу необработанных ягод, воды из необработанных источников. Необходимо тщательно мыть руки, обрабатывать, мыть продукты питания, кипятить воду.
Живаева Е.В.
ФГБОУ ВО «Пермский государственный медицинский университет им. акад. Е.А. Вагнера» Минздрава России
Фрейнд Г.Г.
ФГБОУ ВО «Пермский государственный медицинский университет им. акад. Е.А. Вагнера» Минздрава России
Дизонтогенетические кисты печени: пато- и морфогенез
Журнал: Доказательная гастроэнтерология. 2020;9(3): 39‑46
DOI 10.17116/dokgastro2020903139
Как цитировать
Живаева Е.В., Фрейнд Г.Г. Дизонтогенетические кисты печени: пато- и морфогенез. Доказательная гастроэнтерология. 2020;9(3):39‑46.
Zhivaeva EV, Freynd GG. Dysontogenic liver cysts: patho- and morphogenesis. Russian Journal of Evidence-Based Gastroenterology. 2020;9(3):39‑46. (In Russ.).
2020;9(3):39‑46. (In Russ.).
https://doi.org/10.17116/dokgastro2020903139
Авторы:
Живаева Е.В.
ФГБОУ ВО «Пермский государственный медицинский университет им. акад. Е.А. Вагнера» Минздрава России
Все авторы (2)
Читать метаданные
Представить особенности нарушений эмбриогенеза при различных вариантах дизонтогенетических кист печени.
Врожденные непаразитарные кисты печени встречаются на всем протяжении билиарного тракта, начиная от внутридольковых канальцев и заканчивая интрадуоденальным отделом общего желчного протока. Клинические проявления патологии разнообразны: от полного отсутствия симптомов до печеночной недостаточности, холангита, портальной гипертензии, кровотечения, нагноения кист. В XIX веке появились первые сообщения об обнаружении кист во время оперативных вмешательств или аутопсий. В XX веке с внедрением новых способов визуализации (ультразвукового исследования, компьютерной томографии, магнитно-резонансной томографии) частота обнаружения кист возросла с 0,14 до 7%. В обобщающих описание данной патологии работах представлено несколько классификаций, основанных на этиологии, клинических и макроскопических проявлениях внутрипеченочных кист. На основании анализа морфологических проявлений различных вариантов солитарных кист и поликистоза печени в сопоставлении с этапами нормального эмбриогенеза печени и билиарного тракта сформулированы представления о молекулярных, гормональных и нейроэндокринных аспектах межклеточных взаимодействий паренхиматозных и стромальных элементов ткани печени, нарушения которых ведут к кистообразованию и развитию фиброза печени.
В обобщающих описание данной патологии работах представлено несколько классификаций, основанных на этиологии, клинических и макроскопических проявлениях внутрипеченочных кист. На основании анализа морфологических проявлений различных вариантов солитарных кист и поликистоза печени в сопоставлении с этапами нормального эмбриогенеза печени и билиарного тракта сформулированы представления о молекулярных, гормональных и нейроэндокринных аспектах межклеточных взаимодействий паренхиматозных и стромальных элементов ткани печени, нарушения которых ведут к кистообразованию и развитию фиброза печени.
Изучение публикаций о различных вариантах непаразитарных кист печени свидетельствует о дизонтогенетической природе их возникновения — мальформации дуктальной пластинки, которая происходит с 12-й недели, и миграции бронхиолярных «бутонов» с 3-й по 8-ю неделю эмбрионального развития, это позволяет заключить, что солитарные кисты печени и поликистоз представляют собой достаточно гетерогенную группу заболеваний с различным пато- и морфогенезом.
Ключевые слова:
врожденные кисты печени
поликистоз
реснитчатые переднекишечные печеночные кисты
морфология
Авторы:
Живаева Е.В.
ФГБОУ ВО «Пермский государственный медицинский университет им. акад. Е.А. Вагнера» Минздрава России
Фрейнд Г.Г.
ФГБОУ ВО «Пермский государственный медицинский университет им. акад. Е.А. Вагнера» Минздрава России
Список литературы:
 Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. The American Journal of Pathology. 2008; 172(2):321-332. https://doi.org/10.2353/ajpath.2008.070293
Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. The American Journal of Pathology. 2008; 172(2):321-332. https://doi.org/10.2353/ajpath.2008.070293 1856;7:235.
1856;7:235. 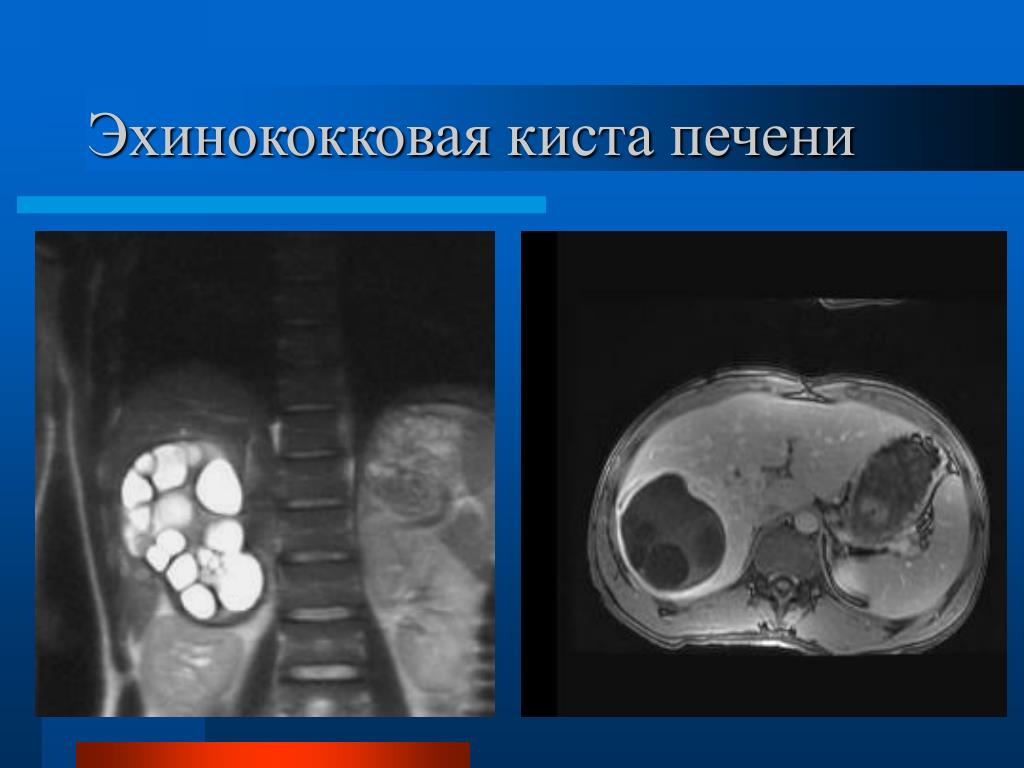 1958;34:488.
1958;34:488.  Laboratory Investigation. 2007;88:112-123. https://doi.org/10.1038/labinvest.3700704
Laboratory Investigation. 2007;88:112-123. https://doi.org/10.1038/labinvest.3700704 Seattle: University of Washington; 2008.
Seattle: University of Washington; 2008.  Human Molecular Genetics. 2008;17:1505-1516. https://doi.org/10.1093/hmg/ddn039
Human Molecular Genetics. 2008;17:1505-1516. https://doi.org/10.1093/hmg/ddn039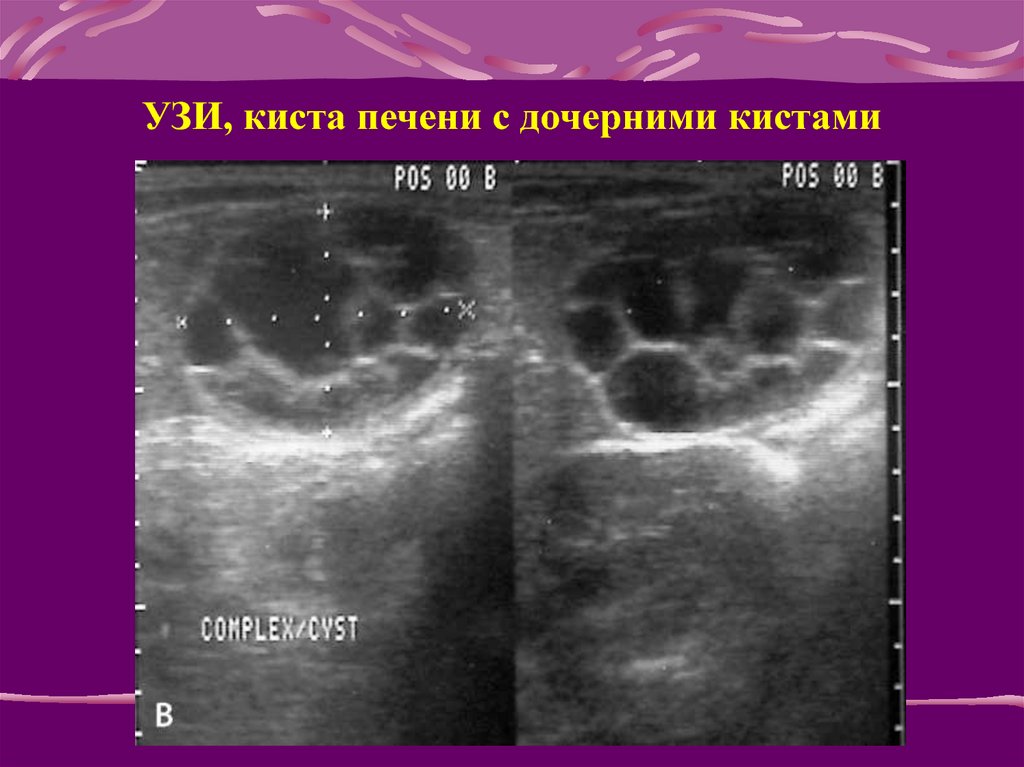 1007/BF00739965
1007/BF00739965 Ciliated hepatic foregut cyst: 103 cases in the world literature. Open Journal of Pathology. 2012;2:45-49. https://doi.org/10.4236/ojpathology.2012.23010
Ciliated hepatic foregut cyst: 103 cases in the world literature. Open Journal of Pathology. 2012;2:45-49. https://doi.org/10.4236/ojpathology.2012.23010 Journal of Cell Biology. 1990;111(2):757-763. https://doi.org/10.1083/jcb.111.2.757
Journal of Cell Biology. 1990;111(2):757-763. https://doi.org/10.1083/jcb.111.2.757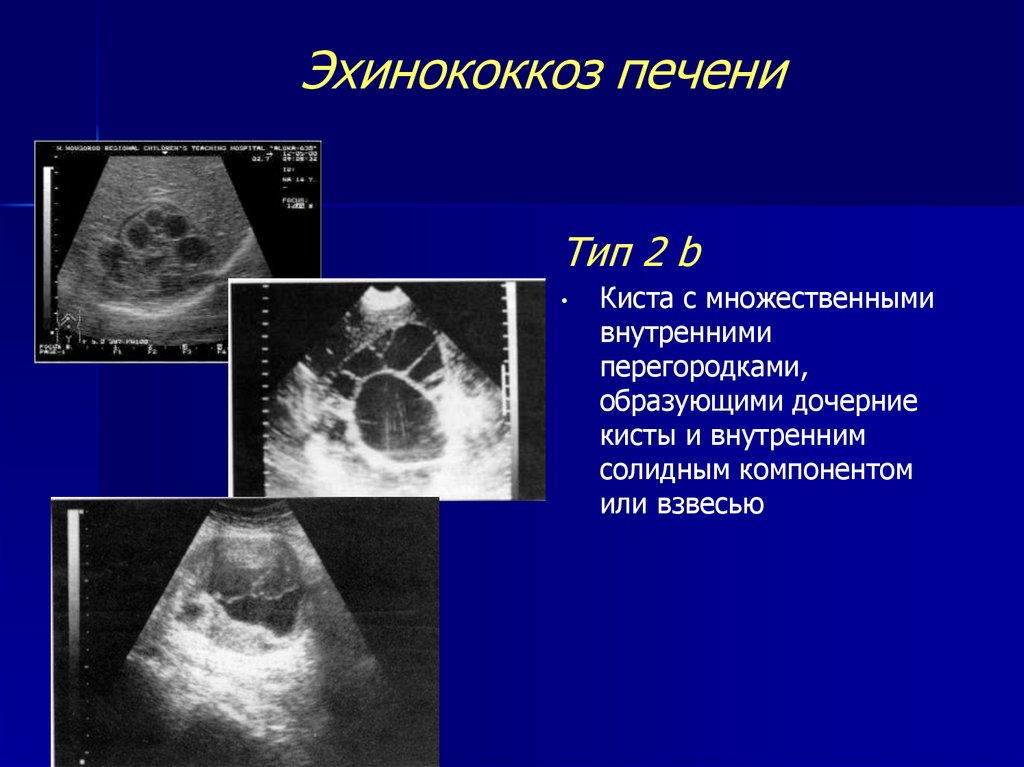 2006;3:377-382. https://doi.org/10.1513/pats.200601-004TK
2006;3:377-382. https://doi.org/10.1513/pats.200601-004TK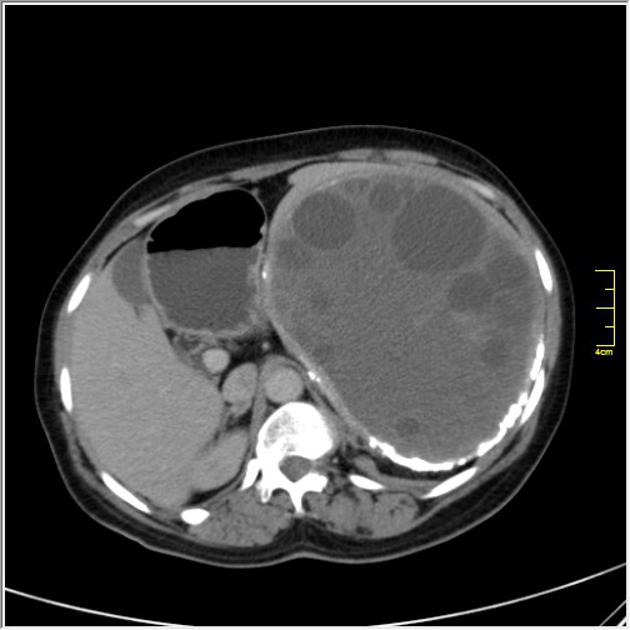 Prenatal and postnatal ciliated hepatic foregut cysts in infants. Journal of Pediatric Surgery. 2010; 45(3):9-14. https://doi.org/10.1016/j.jpedsurg.2009.12.009
Prenatal and postnatal ciliated hepatic foregut cysts in infants. Journal of Pediatric Surgery. 2010; 45(3):9-14. https://doi.org/10.1016/j.jpedsurg.2009.12.009Закрыть метаданные
Врожденные непаразитарные кисты печени встречаются на всем протяжении билиарного тракта, начиная от мельчайших внутридольковых канальцев и заканчивая интрадуоденальной порцией общего желчного протока. Независимо от локализации, клинические проявления кист можно разделить на две большие категории. К первой относятся те, которые зависят от топографии и размеров образований и определяют степень сдавливания окружающих тканей и прилежащих органов. Вторая включает проявления билиарной обструкции различной степени, нередко сопровождающейся холангитом. В редких случаях — при развитии кровоизлияния в полость или разрыве кисты с излиянием содержимого в брюшную полость — возникает картина острого живота, при которой требуется хирургическое вмешательство.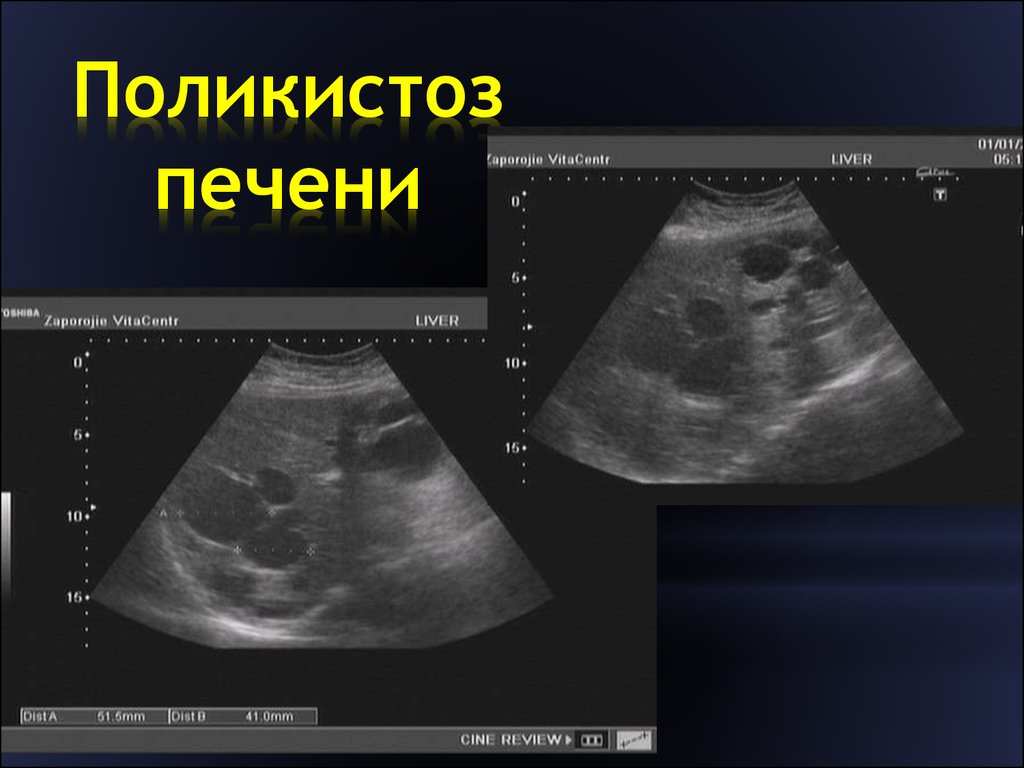 Кисты, возникающие из мелких протоков, не сдавливающие прилежащую ткань печени, зачастую протекают бессимптомно на протяжении многих лет или в течение всей жизни.
Кисты, возникающие из мелких протоков, не сдавливающие прилежащую ткань печени, зачастую протекают бессимптомно на протяжении многих лет или в течение всей жизни.
В начале XX века диагностику внутрипеченочных кист проводили при помощи рентгенографии с применением контраста лишь при наличии клинических симптомов, чем обусловлена редкая выявляемость патологии, которая составляла от 0,14 до 0,17%, по данным хирургических вмешательств и аутопсий [1]. Поэтому в мировой литературе к 1949 г. описано менее 500 случаев различных кист печени, а к 1985 г. — около 900 [2]. До внедрения в практику ультрасонографии правильный диагноз устанавливали лишь у 30% больных, направленных на операции по поводу объемной патологии печени [3]. С широким внедрением в практику неинвазивных инструментальных (ультразвукового исследования, компьютерной томографии, магнитно-резонансной томографии) и серологических методов диагностики увеличилось количество выявленных больных с объемными образованиями печени, а частота выявления кистозных поражений возросла до 7% [4, 5].
Цель обзора — на основании анализа классификаций, результатов генетических и молекулярных исследований, морфологии полостных образований печени представить особенности нарушений эмбриогенеза при различных вариантах дизонтогенетических кист печени.
Первое описание кисты общего желчного протока сделано A.H. Douglas в 1852 г. [6]. В 1865 г. F. Bristowe доложил о случае непаразитарной кистозной болезни печени, обратив внимание на ее сочетание с поликистозной болезнью почек. Наблюдение расценено автором как случайное совпадение, поскольку сочетание кист печени и почек встречалось крайне редко. [7]. Через две недели состоялось заседание общества патологов Лондона, на котором S. Wilks представил аналогичный пример сочетанного кистозного поражения почек и печени, высказав сомнение в том, что данные кисты обусловлены лишь застоем желчи [8]. В 1857 г. N. Freidreich представил описание необычного гистологического типа солитарных кист — foregut hepatic cysts (реснитчатые переднекишечные печеночные кисты — РППК), стенка которых содержала все компоненты стенки кишки, включая мышечный слой [9]. E. Moschcowitz обобщил 85 наблюдений кистозных поражений печени и, проанализировав взгляды различных исследователей на патогенез кистообразования, пришел к выводу, что кисты печени возникают из аберрантных желчных протоков [10]. В 1918 г. H. von Meyenburg описал небольшие группы расширенных мелких желчных протоков в ткани печени при кистозном поражении, которые затем получили название комплексов фон Мейенбурга [11]. В 1955 г. P. Melnick обобщил данные о 70 случаях поликистозной болезни печени за тридцатилетний период. Кисты обнаруживали при аутопсии с частотой 1 на 687 вскрытий [12]. В серии статей о доброкачественных образованиях печени S. Henson и соавт. представили результаты 67 наблюдений одиночных и множественных кист печени: из 38 пациентов с солитарными кистами 12 имели обоснованные показания к хирургическому лечению, у 29 пациентов обнаружен поликистоз при исследовании послеоперационного материала [13, 14]. J. Caroli и соавт. первыми классифицировали состояния печени, при которых наблюдаются кистозные расширения внутрипеченочных желчных протоков, и предложили следующую классификацию кистозных поражений печени:
E. Moschcowitz обобщил 85 наблюдений кистозных поражений печени и, проанализировав взгляды различных исследователей на патогенез кистообразования, пришел к выводу, что кисты печени возникают из аберрантных желчных протоков [10]. В 1918 г. H. von Meyenburg описал небольшие группы расширенных мелких желчных протоков в ткани печени при кистозном поражении, которые затем получили название комплексов фон Мейенбурга [11]. В 1955 г. P. Melnick обобщил данные о 70 случаях поликистозной болезни печени за тридцатилетний период. Кисты обнаруживали при аутопсии с частотой 1 на 687 вскрытий [12]. В серии статей о доброкачественных образованиях печени S. Henson и соавт. представили результаты 67 наблюдений одиночных и множественных кист печени: из 38 пациентов с солитарными кистами 12 имели обоснованные показания к хирургическому лечению, у 29 пациентов обнаружен поликистоз при исследовании послеоперационного материала [13, 14]. J. Caroli и соавт. первыми классифицировали состояния печени, при которых наблюдаются кистозные расширения внутрипеченочных желчных протоков, и предложили следующую классификацию кистозных поражений печени:
1. Истинная поликистозная болезнь печени. Кисты расположены в паренхиме вместе с комплексами фон Мейенбурга, но при этом связи с желчевыводящей системой нет.
Истинная поликистозная болезнь печени. Кисты расположены в паренхиме вместе с комплексами фон Мейенбурга, но при этом связи с желчевыводящей системой нет.
2. Смешанная форма поликистозной болезни. Морфологическая картина та же, но с приступами холангита. Отдельные кисты содержат желчь и сообщаются с билиарным деревом.
3. Кистозные расширения внутрипеченочных желчных протоков. В просветах есть желчь, пациенты страдают от приступов боли и холангита. При этом комплексы фон Мейенбурга отсутствуют. Расширены сегментарные и/или долевые желчные протоки.
4. Врожденное фиброкистозное заболевание. Микрокисты расположены в широких тяжах соединительной ткани (Hamartomes Biliaires Fibreux — желчные гамартомы c фиброзом). При этом нет поражений желчевыделительной системы, но есть признаки портальной гипертензии.
5. Множественные билиарные кисты, ассоциированные с фиброкистозными изменениями, характеризуются наличием портальной гипертензии, кистами в паренхиме и признаками холангита [15].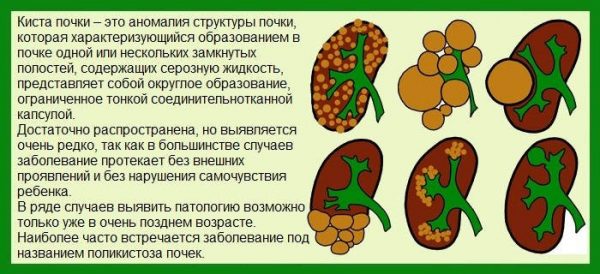
В 1961 г. D. Kerr и соавт. предположили, что врожденный фиброз печени — это состояние, которое связано с врожденными кистами печени и обычно ассоциировано с поликистозом почек [16]. В редакционной статье W. Foulk сообщил, что врожденные аномалии внутрипеченочных желчных протоков у взрослых, при которых диффузно расположенные комплексы фон Мейенбурга обнаруживаются во всей ткани печени, сопровождаются выраженной гипоплазией ветвей портальной вены, и предложил называть такое состояние «врожденным фиброзом печени» [17]. В случае если извитость и дилатация внутрипеченочных желчных протоков являлись доминирующими признаками, а клинические симптомы связаны только с аномалией этих протоков, использовали термин «врожденная дилатация внутрипеченочных желчных протоков». W. Foulk считал, что, хотя комплексы фон Мейенбурга и внутрипеченочный фиброз часто встречаются при поликистозной болезни, последнюю нельзя отождествлять с врожденным фиброзом печени [17].
С начала активного изучения патологии предложено достаточно большое число разнообразных классификаций, в которых, впрочем, есть общие черты: подразделение на солитарные кисты, поликистоз [10, 18, 19], истинные и ложные (травматические) кисты [18, 19]. Клинические классификации учитывают также объем замещенной паренхимы печени, что является определяющим фактором при выборе тактики хирургического лечения [19, 20]. Современной классификацией кист печени можно считать предложенную P. Russo в 2007 г.: паразитарные, одиночные (ретенционные) непаразитарные кисты, реснитчатые переднекишечные печеночные кисты, врожденные (наследственные) кисты печени (врожденный фиброз печени, изолированные дизонтогенетические кисты; аутосомно-доминантное поликистозное заболевание печени и почек; изолированная поликистозная болезнь печени) [21].
Клинические классификации учитывают также объем замещенной паренхимы печени, что является определяющим фактором при выборе тактики хирургического лечения [19, 20]. Современной классификацией кист печени можно считать предложенную P. Russo в 2007 г.: паразитарные, одиночные (ретенционные) непаразитарные кисты, реснитчатые переднекишечные печеночные кисты, врожденные (наследственные) кисты печени (врожденный фиброз печени, изолированные дизонтогенетические кисты; аутосомно-доминантное поликистозное заболевание печени и почек; изолированная поликистозная болезнь печени) [21].
Поликистозные болезни печени — группа генетических болезней, в основном поражающих желчные протоки и нередко эпителий почечных канальцев.
Аутосомно-доминантная поликистозная болезнь почек (АДПБП) является достаточно распространенной врожденной патологией, обнаруживается с частотой от 1:400 до 1:1000 и характеризуется образованием множественных кист в почках, печени и нередко в поджелудочной железе. И хотя синтетическая функция печени, как правило, сохранена, некоторые осложнения кист (кровоизлияние, инфицирование или разрыв), а также их большое количество могут рассматриваться как показание для трансплантации печени.
И хотя синтетическая функция печени, как правило, сохранена, некоторые осложнения кист (кровоизлияние, инфицирование или разрыв), а также их большое количество могут рассматриваться как показание для трансплантации печени.
АРПБП (аутосомно-рецессивная поликистозная болезнь почек) и ассоциированные с печенью фенотипы — болезнь Кароли (БК) и врожденный фиброз печени (ВФП) являются, наоборот, редкими патологиями с распространенностью 1:20 000 новорожденных. БК и ВФП проявляются рецидивирующим острым холангитом и выраженной портальной гипертензией на фоне выраженного перибилиарного фиброза. Описаны также случаи малигнизации. Даже очень редкая изолированная поликистозная болезнь печени (ИПБП) фенотипически аналогична АДПБП, за исключением того, что почки при этом не вовлечены в процесс. Во всех случаях кистозного поражения печени задействован билиарный эпителий, этим обосновано включение данной болезни в группу генетически обусловленных холангиопатий [22].
АДПБП вызывается мутациями одного из двух генов — PKD1 (polycystic kidney disease 1) в 85—90% случаев или PKD2 в 10—15% случаев, — кодирующих соответственно полицистин 1-го типа и полицистин 2-го типа. Полицистины играют роль механо- и хеморецепторов и кальциевых каналов, воспринимают изменения апикального тока [23]. АРПБП/БК и ВФП вызваны мутациями в гене PKHD1 (polycystic kidney and hepatic disease 1), кодирующем фиброцистин — протеин с недостаточно исследованными функциями [23, 24]. И хотя влияние цилиарной дисфункции на физиологию холангиоцита не до конца изучено, животные модели с дефицитом цилиарных белков (полицистина, фиброцистина и апикальных) имеют различные степени билиарной дисгенезии. В печени как АДПБП, так и АРПБП/ВФП/БК морфологически представлены аберрантным состоянием билиарного эпителия, который сохраняет незрелую, протоковидную, пластинчатую архитектонику с последующим формированием билиарных микрогамартом, прогрессивно трансформирующихся в макроскопически различимые кисты, рассеянные по всей паренхиме печени. ИПБП вызвана мутациями в гене PRKCSH (хромосома 19), который кодирует протеин-киназу С, также называемую гепатоцистином, или в гене SEC63 [24].
Полицистины играют роль механо- и хеморецепторов и кальциевых каналов, воспринимают изменения апикального тока [23]. АРПБП/БК и ВФП вызваны мутациями в гене PKHD1 (polycystic kidney and hepatic disease 1), кодирующем фиброцистин — протеин с недостаточно исследованными функциями [23, 24]. И хотя влияние цилиарной дисфункции на физиологию холангиоцита не до конца изучено, животные модели с дефицитом цилиарных белков (полицистина, фиброцистина и апикальных) имеют различные степени билиарной дисгенезии. В печени как АДПБП, так и АРПБП/ВФП/БК морфологически представлены аберрантным состоянием билиарного эпителия, который сохраняет незрелую, протоковидную, пластинчатую архитектонику с последующим формированием билиарных микрогамартом, прогрессивно трансформирующихся в макроскопически различимые кисты, рассеянные по всей паренхиме печени. ИПБП вызвана мутациями в гене PRKCSH (хромосома 19), который кодирует протеин-киназу С, также называемую гепатоцистином, или в гене SEC63 [24]. Ген SEC63 кодирует компонент молекулярной регулировки транслокации и сворачивания синтезированных мембранных гликопротеинов. Гепатоцистин и SEC63 экспрессируются не в ресничках, а в эндоплазматическом ретикулуме, вследствие этого кистозные болезни печени могут быть вызваны дефектами синтеза протеинов не только ресничками [25, 26].
Ген SEC63 кодирует компонент молекулярной регулировки транслокации и сворачивания синтезированных мембранных гликопротеинов. Гепатоцистин и SEC63 экспрессируются не в ресничках, а в эндоплазматическом ретикулуме, вследствие этого кистозные болезни печени могут быть вызваны дефектами синтеза протеинов не только ресничками [25, 26].
В эмбриогенезе ткань печени и билиарная система развиваются из переднего отдела первичной кишечной трубки. Изначально субпопуляция клеток-предшественников, рассматриваемых в качестве предшественников эпителиальных клеток желчных протоков и гепатоцитов, локализуется вблизи мезенхимы формирующихся портальных сосудов, которая, в свою очередь, экспрессирует желчеспецифические цитокератины [7, 19]. С 8-й недели слой клеток-предшественников формирует непрерывную однослойную кольцевую структуру, называемую первичной дуктальной пластинкой, и окружающую портальную мезенхиму. С 12-й недели эмбрионального развития дуктальная пластинка частично становится двухслойной.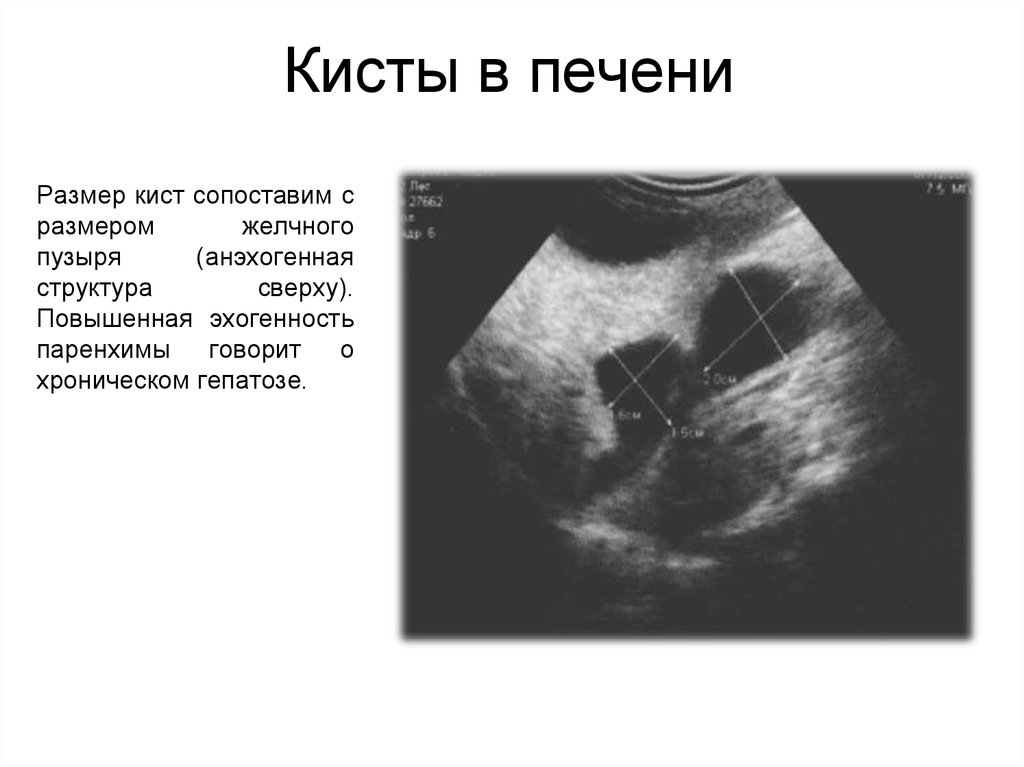 Локальные расширения дуктальной пластинки в местах, где она двухслойная, дают начало желчным протокам [27]. Оставшаяся часть дуктальной пластинки в дальнейшем регрессирует. Ремоделирование дуктальной пластинки во время фетального и постнатального развития — динамичный процесс пролиферации и апоптоза клеток. Угнетение созревания и неполноценное ремоделирование приводят к избыточному количеству незрелых эмбриональных билиарных структур, нарушение инволюции которых является одной из причин формирования кист печени [28]. Однако у людей кисты могут формироваться и на протяжении всей жизни. У мышей с условным генным нокаутом PKD1 или PKD2 наблюдается прогрессирующее образование кист печени и почек, напоминающее болезни человека, даже когда индукция осуществляется через несколько недель после рождения [29—31]. Это указывает на то, что полицистин играет важную роль в поддержании нормальной билиарной архитектоники во взрослой жизни.
Локальные расширения дуктальной пластинки в местах, где она двухслойная, дают начало желчным протокам [27]. Оставшаяся часть дуктальной пластинки в дальнейшем регрессирует. Ремоделирование дуктальной пластинки во время фетального и постнатального развития — динамичный процесс пролиферации и апоптоза клеток. Угнетение созревания и неполноценное ремоделирование приводят к избыточному количеству незрелых эмбриональных билиарных структур, нарушение инволюции которых является одной из причин формирования кист печени [28]. Однако у людей кисты могут формироваться и на протяжении всей жизни. У мышей с условным генным нокаутом PKD1 или PKD2 наблюдается прогрессирующее образование кист печени и почек, напоминающее болезни человека, даже когда индукция осуществляется через несколько недель после рождения [29—31]. Это указывает на то, что полицистин играет важную роль в поддержании нормальной билиарной архитектоники во взрослой жизни.
Персистирование несформированных дуктальных элементов стимулирует фиброгенез в портальных трактах печени, развивается вторичный холангит или портальная гипертензия и связанные с ней осложнения. Длительно существующая портальная гипертензия может приводить к вторичному тромбозу портальных вен и к кавернозной трансформации портальных вен (КТПВ) в итоге. Существует точка зрения, что КТПВ также является одним из проявлений нарушений эмбриогенеза [32]. Необходимо учитывать, что развитие желчных протоков и васкуляризация печени — взаимосвязанные процессы. Установлено, что мальформация дуктальной пластинки ассоциирована с мальформацией портальных вен по типу «безвершинной ивы», для которой характерно наличие большого числа тесно расположенных в портальных трактах венул [33]. В зависимости от выраженности нарушений эмбриогенеза в процесс могут быть вовлечены как мелкие междольковые желчные протоки — врожденный фиброз печени, так и средние внутрипеченочные протоки — БК. Одновременный патологический процесс в обеих структурах обозначается как синдром Кароли [34]. В зависимости от особенностей дизэмбриогенеза наблюдаются морфогенетические различия между АДПБП и АРПБП. При АДПБП зарождающиеся кисты отделяются от исходного протока и образуют автономные структуры, которые не связаны с желчным деревом; при АРПБП сообщение между кистами и желчными протоками, как правило, сохраняется.
Длительно существующая портальная гипертензия может приводить к вторичному тромбозу портальных вен и к кавернозной трансформации портальных вен (КТПВ) в итоге. Существует точка зрения, что КТПВ также является одним из проявлений нарушений эмбриогенеза [32]. Необходимо учитывать, что развитие желчных протоков и васкуляризация печени — взаимосвязанные процессы. Установлено, что мальформация дуктальной пластинки ассоциирована с мальформацией портальных вен по типу «безвершинной ивы», для которой характерно наличие большого числа тесно расположенных в портальных трактах венул [33]. В зависимости от выраженности нарушений эмбриогенеза в процесс могут быть вовлечены как мелкие междольковые желчные протоки — врожденный фиброз печени, так и средние внутрипеченочные протоки — БК. Одновременный патологический процесс в обеих структурах обозначается как синдром Кароли [34]. В зависимости от особенностей дизэмбриогенеза наблюдаются морфогенетические различия между АДПБП и АРПБП. При АДПБП зарождающиеся кисты отделяются от исходного протока и образуют автономные структуры, которые не связаны с желчным деревом; при АРПБП сообщение между кистами и желчными протоками, как правило, сохраняется. Этим обусловлены и различные клинические проявления при АРПБП, ВФП, БК и АДПБП.
Этим обусловлены и различные клинические проявления при АРПБП, ВФП, БК и АДПБП.
До настоящего момента гистогенез РППК не изучен детально. Согласно литературным данным, кисты могут возникать из эмбрионального зачатка передней кишки (из которого развиваются ткани ротоглотки, пищевод, желудок, двенадцатиперстная кишка, печень, желчный пузырь, поджелудочная железа, трахеобронхиальное дерево и легкие), простирающегося от ротоглотки до печеночного дивертикула [35, 36]. Нижний отдел респираторного тракта начинает формироваться в конце 3-й недели эмбрионального развития в виде дивертикула вентрального краниального зачатка, из которого развиваются трахея, бронхиальное дерево и легочные почки. Печеночный зачаток начинает дифференцироваться на 4-й неделе и развивается из энтодермальных клеток, которые пролиферируют в поперечной перегородке и становятся гепатоцитами. Участок мезодермы между перикардиальной полостью и пупочно-кишечным протоком, называемый поперечной перегородкой, участвует в разделении грудной и брюшной полостей, которое является неполным.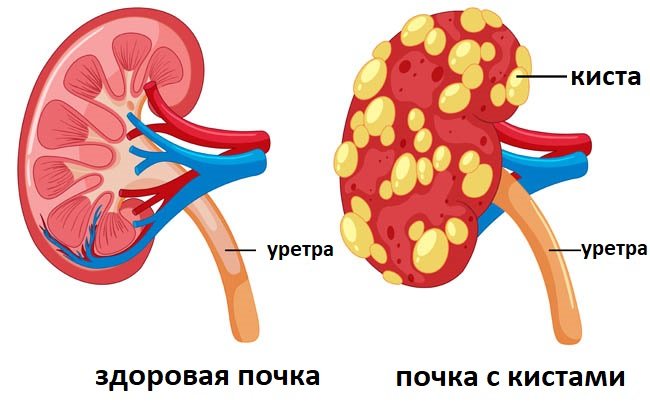 Благодаря этому возникают плевроперитонеальные каналы, которые позволяют расти легочным зачаткам. Закрытие данных каналов происходит на 8-й неделе из-за слияния плевроперитонеальной мембраны с поперечной перегородкой.
Благодаря этому возникают плевроперитонеальные каналы, которые позволяют расти легочным зачаткам. Закрытие данных каналов происходит на 8-й неделе из-за слияния плевроперитонеальной мембраны с поперечной перегородкой.
Зачатки бронхиол имеют возможность мигрировать из грудной полости в брюшную через плевроперитонеальный канал до момента его закрытия. Такие аномальные бронхиолярные «бутоны» могут быть включены в поперечную перегородку, а затем окружены энтодермальными клетками печеночного дивертикула [37]. В процессе внутриутробного развития левая печеночная доля имеет большую массу. Ремоделирование органа с увеличением правой доли и регрессией левой начинается на 6—8-й неделе, что может объяснить преимущественную локализацию РППК в левой доле [38]. Начиная с 10-й недели цилиндрический эпителий, формирующий бронхиальную почку, может быть замещен респираторным эпителием с реснитчатыми клетками, слизистыми клетками, эндокринными клетками и клетками Клара, что подтверждается иммуногистохимическими методами исследования [39]. Изучение внутрибрюшных бронхогенных кист подтвердило гипотезу миграции аномальных бронхиолярных «бутонов», но, в отличие от бронхогенной кисты, в стенке РППК никогда не содержится хрящ [39—41].
Изучение внутрибрюшных бронхогенных кист подтвердило гипотезу миграции аномальных бронхиолярных «бутонов», но, в отличие от бронхогенной кисты, в стенке РППК никогда не содержится хрящ [39—41].
Врожденные кисты печени часто сочетаются с фиброзом. Ряд исследователей пытались выяснить патофизиологические механизмы, лежащие в основе неправильного и избыточного фиброзного ответа при ВФП. Звездчатые клетки печени (ЗКП) играют ключевую роль в развитии фиброза при заболеваниях печени, в том числе и при врожденном фиброзе печени [42, 43]. Деградация базальной мембраны и компонентов экстрацеллюлярного матрикса, а также их ремоделирование играют важную роль в пре- и постнатальном периодах развития. Компоненты базальной пластинки, такие как ламинин и коллаген IV типа, наряду с координированной экспрессией протеолитических энзимов играют важную роль в нормальном развитии внутрипеченочных желчных протоков. Большинство протеолитических энзимов, вовлеченных в эти процессы, принадлежат к классу матричных металлопротеиназ (ММП) и сериновых протеиназ, в особенности активаторов плазминогена (АП)/плазминовой системы. Известно, что плазмин способствует активации ММП-9 и ММП-13, которые играют важную роль в деградации компонентов базальной мембраны, в том числе коллагена IV типа. W. Sweeney и E. Avner считают, что билиарная гиперэкспрессия плазминогена и АП ведет к генерации избыточных количеств плазмина и последующему плазмин-зависимому лизису молекул экстрацеллюлярного матрикса, что может способствовать билиарной дисгенезии при врожденном фиброзе печени [44].
Известно, что плазмин способствует активации ММП-9 и ММП-13, которые играют важную роль в деградации компонентов базальной мембраны, в том числе коллагена IV типа. W. Sweeney и E. Avner считают, что билиарная гиперэкспрессия плазминогена и АП ведет к генерации избыточных количеств плазмина и последующему плазмин-зависимому лизису молекул экстрацеллюлярного матрикса, что может способствовать билиарной дисгенезии при врожденном фиброзе печени [44].
Гиперэкспрессия гена остеопонина также является важным компонентом патогенеза различных вариантов билиарной атрезии и врожденных холестатических синдромов — врожденного фиброза печени и БК. Остеопонин — стимулятор воспаления, и его избыточный синтез регулируется наличием избыточных количеств регуляторных факторов (NF-κB и ТФР-β1) [45].
В работе, посвященной изучению связи избыточного количества малодифференцированных желчных протоков и фиброза, Y. Sato и соавт. на модели крысы продемонстрировали, что в присутствии ТФР-β1 холангиоциты приобретают черты мезенхимальных клеток, уподобляясь фибробластам, т. е. происходит эпителиально-мезенхимальный переход (ЭМП). Авторы предположили, что гиперэкспрессия молекул экстрацеллюлярного матрикса этими клетками может быть причиной прогрессирования фиброза печени [46].
е. происходит эпителиально-мезенхимальный переход (ЭМП). Авторы предположили, что гиперэкспрессия молекул экстрацеллюлярного матрикса этими клетками может быть причиной прогрессирования фиброза печени [46].
Некоторые экспериментальные и клинические исследования свидетельствуют о том, что пролиферирующие холангиоциты активно откликаются на гормоны, нейропептиды, факторы роста, цито- и хемокины, секретируя, в свою очередь, большое количество различных агентов, которые через аутокринные механизмы регуляции подкрепляют систему пролиферации, тормозят апоптоз и способствуют «избыточной» коммуникации с другими типами клеток печени (гепатоцитами, звездчатыми и эндотелиальными клетками) [47]. Среди этих веществ эстрогены и инсулиноподобный фактор роста 1-го типа играют решающую роль в поддержании пролиферативной активности холангиоцитов крысы и человека, воздействуя на специфические рецепторы. Они усиливают пролиферативный эффект, оказываемый на рецепторный и пострецепторный уровни, с помощью которых активируются «пути выживания». «Реактивные» или «активированные» холангиоциты формируются из прогениторных клеточных компартментов, расположенных максимально близко к терминальным холангиолам в канальцах Геринга. Они могут инициировать пролиферацию воспалительных и мезенхимальных клеток и синтез последними компонентов экстрацеллюлярного матрикса [48]. Хотя мезенхимальные клетки считаются реализаторами фиброза, реактивные холангиоциты называют «водителями ритма фиброза печени» [49]. Список факторов, на основе которых происходит межклеточное взаимодействие, постоянно расширяется: интерлейкин-6, интерлейкин-8, фактор некроза опухоли α, гамма-интерферон, моноцитарный хемотаксический фактор 1-го типа, цитокин-индуцированный хемоаттрактант нейтрофилов и оксид азота, который регулирует иммунную активность лимфоцитов и полиморфноядерных клеток. Активированные холангиоциты также синтезируют фактор роста эндотелия сосудов, эндотелин-1, тромбоцитарный фактор роста ВВ, трансформирующий фактор роста β2 и фактор роста фибробластов.
«Реактивные» или «активированные» холангиоциты формируются из прогениторных клеточных компартментов, расположенных максимально близко к терминальным холангиолам в канальцах Геринга. Они могут инициировать пролиферацию воспалительных и мезенхимальных клеток и синтез последними компонентов экстрацеллюлярного матрикса [48]. Хотя мезенхимальные клетки считаются реализаторами фиброза, реактивные холангиоциты называют «водителями ритма фиброза печени» [49]. Список факторов, на основе которых происходит межклеточное взаимодействие, постоянно расширяется: интерлейкин-6, интерлейкин-8, фактор некроза опухоли α, гамма-интерферон, моноцитарный хемотаксический фактор 1-го типа, цитокин-индуцированный хемоаттрактант нейтрофилов и оксид азота, который регулирует иммунную активность лимфоцитов и полиморфноядерных клеток. Активированные холангиоциты также синтезируют фактор роста эндотелия сосудов, эндотелин-1, тромбоцитарный фактор роста ВВ, трансформирующий фактор роста β2 и фактор роста фибробластов.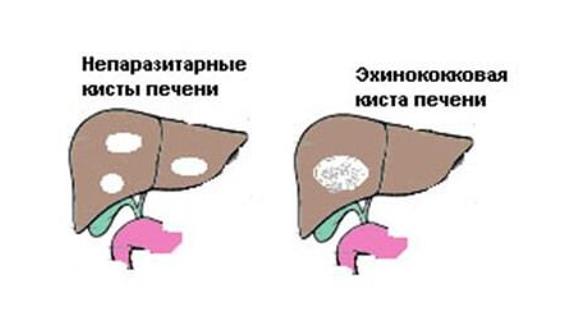
Помимо установления паракринной связи с мезенхимными клетками холангиоциты могут участвовать в генерации фиброза печени через ЭМП. ЭМП — это процесс клеточного перепрограммирования, при котором эпителиальные клетки приобретают некоторые фенотипические и функциональные характеристики мезенхимальных клеток, такие как экспрессия фибробласт-специфических маркеров (продуктов деградации фибрина-1, виментина), способность мигрировать путем локального демонтажа основания мембраны, на которой находится эпителиальный слой, и способность генерировать различные компоненты соединительной ткани (фибронектин, коллаген, эластин, тенасцин). Таким образом, ЭМП может способствовать накоплению активированных фибробластов в связи с потерей желчных протоков. Этот биологический процесс также описан в патогенезе фиброза органов в почках [50] и легких [51]. Недавние исследования показывают, что ЭМП также вовлечен в фиброз печени за счет активного синтеза ТФР-β2 ЗКП [22, 52]. Активация ЗКП первично инициируется ТФР-β1, полученным из клеток Купфера. Принято считать, что ТФР-β2 — сильнодействующий ингибитор роста и профибротический цитокин, который играет основную роль в физиологических процессах заживления ран и патогенезе фиброза в различных органах. В патологических условиях это приводит к аккумуляции фиброзного матрикса, а в процессе репаративной регенерации — к оптимальному формированию рубцовой ткани [48, 49].
Принято считать, что ТФР-β2 — сильнодействующий ингибитор роста и профибротический цитокин, который играет основную роль в физиологических процессах заживления ран и патогенезе фиброза в различных органах. В патологических условиях это приводит к аккумуляции фиброзного матрикса, а в процессе репаративной регенерации — к оптимальному формированию рубцовой ткани [48, 49].
В ответ на повреждение реактивные холангиоциты приобретают фенотип, сходный с нейроэндокринными клетками, и получают возможность отвечать на нейроэндокринные стимулы. Клетки получают β1 и β2 адренергические рецепторы, М3 ацетилхолиновые рецепторы [53], рецепторы серотонина 1A и 1B [54]. В случае холестаза холангиоциты могут также непосредственно секретировать серотонин, ограничивая рост желчных протоков с помощью дополнительной аутокринной ингибиторной петли.
Морфологическая картина как при солитарных кистах, так и при поликистозе печени представлена фиброзной капсулой различной толщины, зачастую с ангиоматозом, наличием малодифференцированных билиарных структур и островков гепатоцитов. Однослойный кубический эпителий билиарного типа или призматический эпителий выстилает кисты [55]. Строение стенки РППК отличается от простых солитарных кист. Их стенка четырехслойная: цилиндрический мерцательный эпителий, рыхлая соединительная ткань, пучки гладкомышечных клеток различной толщины, нервные стволики и фиброзная капсула [38, 39, 55—57]. Наличие ресничек подтверждается электронной микроскопией. Содержимое кист может представлять собой слизь, желчь или серозную жидкость. В случае нагноения в толще стенки и просветах кисты отмечаются скопления нейтрофильных гранулоцитов, макрофагов и колоний микроорганизмов [55—57].
Однослойный кубический эпителий билиарного типа или призматический эпителий выстилает кисты [55]. Строение стенки РППК отличается от простых солитарных кист. Их стенка четырехслойная: цилиндрический мерцательный эпителий, рыхлая соединительная ткань, пучки гладкомышечных клеток различной толщины, нервные стволики и фиброзная капсула [38, 39, 55—57]. Наличие ресничек подтверждается электронной микроскопией. Содержимое кист может представлять собой слизь, желчь или серозную жидкость. В случае нагноения в толще стенки и просветах кисты отмечаются скопления нейтрофильных гранулоцитов, макрофагов и колоний микроорганизмов [55—57].
Кровоизлияния в стенках кист могут быть обусловлены не только оперативным вмешательством, связанным со способом удаления и обработки кист, но и наличием сосудов с явными признаками дисплазии в их стенках [39—41, 55—57]. В стенке возможно формирование многорядных структур с пролиферацией, представленных малодифференцированными (так называемыми резервными) клетками.
В ткани печени отмечаются дистрофия гепатоцитов, внутриклеточный или каналикулярный холестаз, умеренный или выраженный фиброз портальных трактов, капилляризация синусоидов, перивенулярный склероз, что обусловливает развитие портальной гипертензии. Более того, зачастую выявляются малодифференцированные желчные структуры, частично кистозно расширенные. Междольковые артериолы и венулы часто формируют конгломераты в портальных трактах. Описанные изменения могут иметь как врожденную, так и приобретенную природу. Врожденные изменения характеризуются дисплазией различных структур ткани печени — гипоплазией долек, дисплазией венозных и артериальных сосудов и желчных протоков. В стенках кист также отмечаются признаки тканевой дисплазии — незрелые билиарные структуры (комплексы Мейенбурга), островки гепатоцитов, конгломераты сосудов артериального и венозного типов [55, 57]. Часто встречаются тяжи клеток билиарного эпителия, состоящие из персистирующих элементов эмбриональной дуктальной пластинки и скопления малодифференцированных клеток билиарного эпителия [55, 57].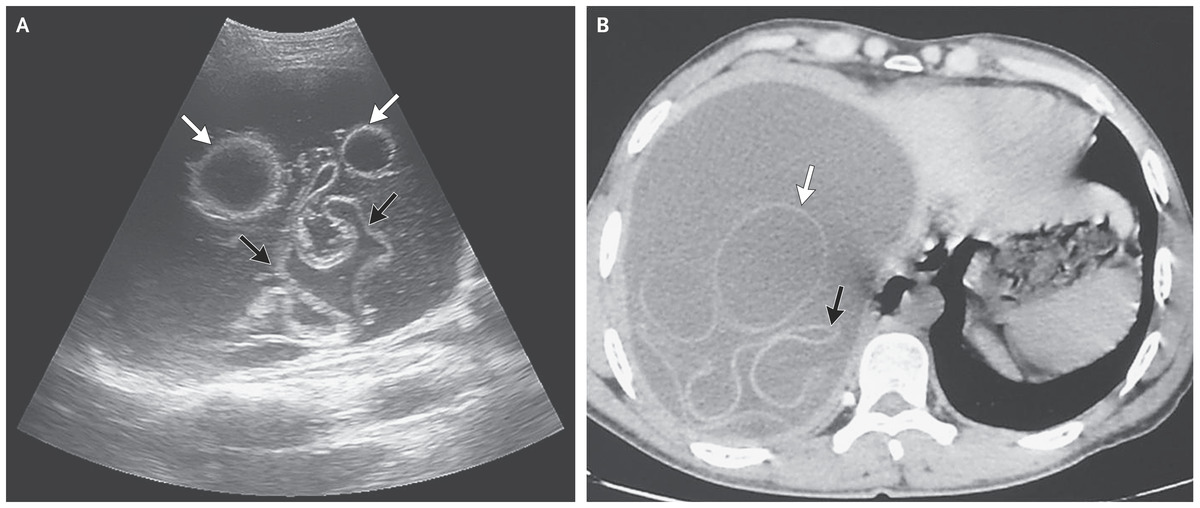
Анализ публикаций, посвященных изучению непаразитарных кист печени, позволяет заключить, что кисты печени имеют дизонтогенетическую природу и представляют собой достаточно гетерогенную группу заболеваний с различным пато- и морфогенезом. Поликистоз и солитарные кисты, содержащие фиброзную капсулу и выстилку уплощенным билиарным или многорядным эпителием, следует рассматривать как мальформации дуктальной пластинки. Гистологические и иммуногистохимические особенности реснитчатых переднекишечных печеночных кист сходны с таковыми в бронхиолах нижних дыхательных путей, что подтверждает их происхождение из трахеобронхиального зачатка, являющегося, как и пузырно-печеночный дивертикул, производным первичной кишки.
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflict of interest.
Кисты - почки; Почки - поликистозные; Аутосомно-доминантный поликистоз почек; АДПКД
Поликистоз почек (PKD) — заболевание почек, передающееся по наследству. При этом заболевании в почках образуется много кист, что приводит к их увеличению.
При этом заболевании в почках образуется много кист, что приводит к их увеличению.
КТ брюшной полости показывает кисты в печени и почках (поликистоз). Печень — большой орган в левой части экрана. Темные пятна в печени – это кисты.
КТ брюшной полости показывает множественные кисты в печени и селезенке. Обратите внимание на темную круглую кисту в печени (левая часть экрана) и большую круглую кисту неправильной формы в селезенке (внизу, справа на экране).
ПКП передается по наследству. Две наследуемые формы поликистозной болезни — аутосомно-доминантная и аутосомно-рецессивная.
У людей с поликистозом почек имеется множество скоплений кист в почках. Что именно вызывает образование кист, неизвестно.
PKD связана со следующими состояниями:
Около половины людей с поликистозом почек имеют кисты в печени.
Симптомы поликистозной болезни почек могут включать любые из следующих:
Обуждение может показать:

Анализы, которые могут быть выполнены, включают:
Людям с головными болями в личном или семейном анамнезе поликистозов необходимо пройти обследование, чтобы определить, не являются ли причиной церебральные аневризмы.
Поликистозная болезнь почек и кисты печени или других органов могут быть обнаружены с помощью следующих тестов:
Целью лечения является контроль симптомов и предотвращение осложнений.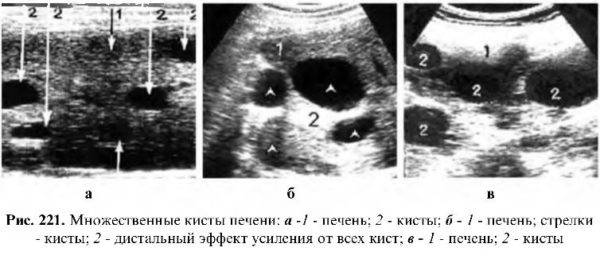 Лечение может включать:
Лечение может включать:
Любую инфекцию мочевыводящих путей следует быстро лечить антибиотиками.
Болезненные, инфицированные, кровоточащие или вызывающие закупорку кисты могут потребовать дренирования. Обычно кист слишком много, чтобы было целесообразно удалить каждую кисту.
Может потребоваться операция по удалению одной или обеих почек. Лечение терминальной стадии заболевания почек может включать диализ или трансплантацию почки.
Дополнительную информацию и поддержку людям с поликистозом почек и их семьям можно найти в группе поддержки при заболеваниях почек.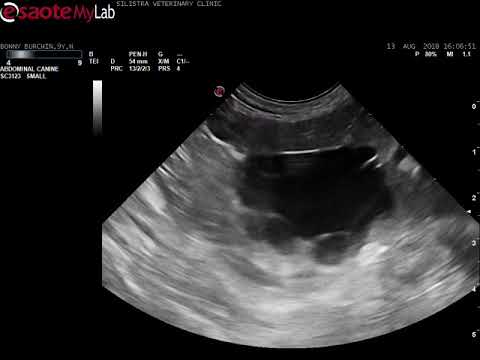
Болезнь ухудшается медленно. В конечном итоге это может привести к терминальной стадии почечной недостаточности. Это также связано с заболеванием печени, включая инфекцию кист печени.
Лечение может облегчить симптомы на многие годы.
Люди с поликистозом почек, у которых нет других заболеваний, могут быть хорошими кандидатами на трансплантацию почки.
9Обратитесь к своему поставщику медицинских услуг, если:
В настоящее время никакое лечение не может предотвратить образование или увеличение кист.
Арнаут М.А. Кистозные заболевания почек. В: Goldman L, Schafer AI, ред. Медицина Голдман-Сесил . 26-е изд. Филадельфия, Пенсильвания: Elsevier; 2020: глава 118.
Торрес В.Е., Харрис ПК. Кистозные заболевания почек. В: Ю. А.С.Л., Чертоу Г.М., Луккс В.А., Марсден П.А., Скорецки К., Таал М.В., ред. Бреннер и Ректор Почка . 11-е изд. Филадельфия, Пенсильвания: Elsevier; 2020: глава 45.
Последнее рассмотрение: 27.07.2021
Рецензировал: Валеад Латиф, доктор медицинских наук, нефролог и клинический доцент Медицинской школы Рутгерса, Ньюарк, Нью-Джерси. Обзор предоставлен VeriMed Healthcare Network. Также рассмотрены Дэвидом Зивом, доктором медицины, MHA, медицинским директором, Брендой Конауэй, редакционным директором, и A.D.A.M. Редакционная коллегия.
Аутосомно-доминантный поликистоз почек (АДПБП) иногда может приводить к потенциально серьезным осложнениям в других частях тела, помимо почек.
У многих людей с АДПБП развиваются кисты в других органах, а также в почках. Печень также часто поражается ADPKD.
Кисты, развивающиеся в печени, обычно не нарушают нормальную функцию печени, но иногда они могут инфицироваться или вызывать такие симптомы, как:
В большинстве случаев эти симптомы проходят без необходимости лечения .
В редких случаях, когда большая киста вызывает сильную или постоянную боль, может потребоваться хирургическое вмешательство для дренирования кисты.
В очень редких случаях печень может настолько сильно разбухнуть, что перестает работать должным образом.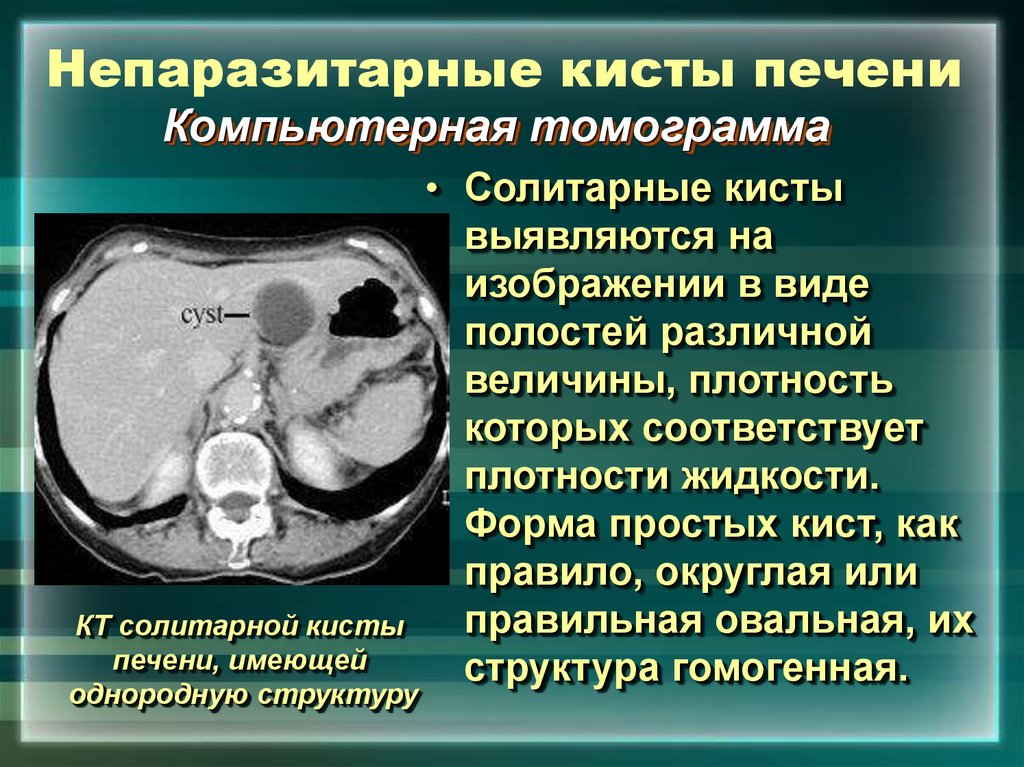
В таких случаях может потребоваться хирургическое удаление части печени или полная пересадка печени.
В результате высокого кровяного давления у людей с АДПБП также повышен риск развития сердечно-сосудистых заболеваний (ССЗ).
ССЗ — это общий термин, который относится к состояниям, поражающим сердце и кровеносные сосуды, и включает:

Внесение изменений в здоровый образ жизни, таких как отказ от курения, снижение потребления алкоголя, регулярные физические упражнения и здоровое сбалансированное питание, также может снизить риск развития сердечно-сосудистых заболеваний.
Узнайте больше о профилактике сердечно-сосудистых заболеваний
Аневризма представляет собой выпячивание кровеносного сосуда, вызванное слабостью стенки кровеносного сосуда.
Когда кровь проходит через ослабленную часть сосуда, кровяное давление заставляет его выпячиваться наружу, как воздушный шар.
Аневризмы головного мозга чаще встречаются у людей с АДПБП, чем у населения в целом, вероятно, потому, что высокое кровяное давление влияет на ослабленные стенки кровеносных сосудов.
Аневризма головного мозга обычно не вызывает каких-либо заметных симптомов, если только она не лопнет (разорвется).
Разрыв аневризмы вызывает кровотечение на поверхности головного мозга. Это известно как субарахноидальное кровоизлияние.
Симптомы субарахноидального кровоизлияния могут включать:
Субарахноидальное кровоизлияние является неотложной медицинской помощью, требующей немедленного лечения для предотвращения серьезных осложнений, повреждения головного мозга и смерти.
Немедленно наберите 999 и вызовите скорую помощь, если вы считаете, что у вас или у кого-то из ваших знакомых субарахноидальное кровоизлияние.
По оценкам, примерно у 10% людей с АДПБП разовьется аневризма головного мозга, но у большинства из них не будет никаких симптомов, и это никогда не вызовет проблем.
Люди с ADKPD, у которых также есть семейный анамнез субарахноидальных кровоизлияний, подвергаются большему риску.
Если у вас АДПБП и в семейном анамнезе были субарахноидальные кровоизлияния, вам обычно предложат МРА-сканирование для проверки наличия аневризм в головном мозге.
МРА-сканирование использует магнитное поле и радиоволны для получения изображений артерий и кровотока в них.
Если аневризмы не обнаружены или обнаружены только небольшие аневризмы, вам будет предложено дальнейшее сканирование с интервалом от 1 до 5 лет для проверки наличия новых кровоизлияний или увеличения размера уже существующего.