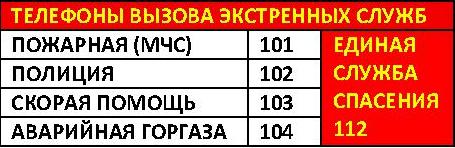







2011-2017 © МБУЗ ГКП № 7, г.Челябинск.
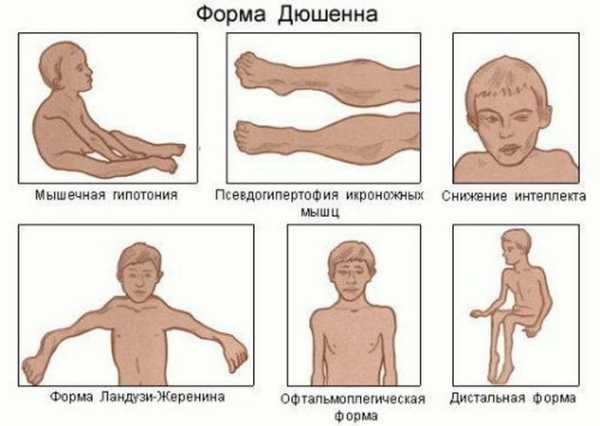
Миодистрофия Дюшенна – наследственное нервно-мышечное заболевание, которое проявляется у мальчиков и характеризуется прогрессирующей мышечной слабостью и утомляемостью, симметричными псевдогипертрофиями голеней и симметричными атрофиями других мышц в сочетании с костно-суставными деформациями, сердечно-сосудистыми и дыхательными нарушениями. Миодистрофия Дюшенна имеет Х-сцепленный рецессивный тип наследования, Английской аббревиатурой DMD обозначается как название гена дистрофина, так и название заболевания – мышечная дистрофия Дюшенна.
Ген дистрофина DMD локализован на коротком плече Х-хромосомы (локус Xp21.2–р21.1). Это один из самых больших генов человека, поэтому в нем так часто возникают спорадические мутации (до 40%). Заболевание в 60% случаев наследуется мальчиками от женщин-носительниц, как типичное Х-сцепленное рецессивное наследование.
Поэтому мы часто говорим, миодистрофия Дюшенна– болезнь мальчиков. У женщин 2 Х-хромосомы и в случае, если возникнет поломка гена на одной хромосоме, вторая будет вырабатывать дистрофин.
Отсутствие дистрофина (так происходит при мышечной дистрофии Дюшенна) влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Патогенез миодистрофии Дюшенна, включает и иммунопатологические процессы. В организме нарушен процесс регенерации и почти сразу после рождения запускается каскад воспалительной реакции. Из-за возникающего дефекта липидного слоя сарколеммы повышается ее проницаемость, что провоцирует рабдомиолиз. Через отсутствующий защитный барьер клетки внутриклеточная КФК выходит в кровь, а внеклеточный кальций в миоциты.
Выделяют 5 стадий заболевания.
| Стадия | Описание, признаки | |
|---|---|---|
| 1 | Досимптомная | Повышение уровней КФК, ЛДГ, АСТ и АЛТ в биохимическом анализе крови. |
| 2 | Ранняя (амбулаторная) Сохранена способность к самостоятельному передвижению | Использование приёмов Говерса при вставании, миопатическая «утиная» походка, ходьба на носках. Ребёнок поднимается по ступенькам приставным шагом и\или с поддержкой. |
| 3 | Поздняя (амбулаторная) Сохранена способность к самостоятельному передвижению | Нарастающие трудности при ходьбе, потеря способности подниматься по ступенькам и вставать с пола. |
| 4 | Ранняя (не амбулаторная) Утрачена способность к самостоятельному передвижению | Пациент способен некоторое время передвигаться самостоятельно, способен удерживать положение тела. Высокие риски развития скелетных деформаций, кардиомиопатии и дыхательных нарушений. |
| 5 | Поздняя (не амбулаторная) Утрачена способность к самостоятельному передвижению | Нарастание ограничения функций верхних конечностей, скелетных деформаций, кардиомиопатии и дыхательных нарушений. Трудности с удержанием положения тела. |
Новорожденные с миодистрофией Дюшенна могут не иметь значительных или заметных отклонений. Тогда, первые месяцы и даже год моторное развитие ребенка происходит в пределах нормы или с незначительной задержкой. До 30% пациентов на первом году жизни имеют отставание в психоречевом развитии. Также у таких детей, чаще, чем в среднем в популяции, выявляются расстройства аутистического спектра.
До 10% пациентов с ПМД Дюшенна могут иметь клинические проявления на 1-м году жизни в виде симптомокомплекса «вялого ребенка». Основными симптомами являются слабость мышц тазового пояса, патологическая мышечная утомляемость при физической нагрузке. Изменение походки по типу «утиной» – к 4-5 годам формируется дефект походки: больной широко расставляет ноги, при ходьбе переваливается из стороны в сторону, передвигается на носочках, пациенты помогают себе руками, сильно размахивая ими при ходьбе. В среднем пациенты с миопатией Дюшенна теряют способность самостоятельно передвигаться с 8 до 12 лет. Однако бывают потерявшие способность к самостоятельному передвижению пациенты и в 6 лет, и пациенты, самостоятельно передвигающиеся в 16-17 лет. По всей видимости, это зависит от индивидуальных особенностей течения заболевания.
Сердечно-сосудистые расстройства — лабильность пульса, АД, приглушение тонов, расширение границ сердца, сердечная недостаточность. Примерно 73% популяции имеют проявления кардиальной патологии. Сердечно-сосудистая система вовлекается в патологический процесс достаточно рано. Отмечаются изменения миокарда (блокада ножек пучка Гиса и др.).
Первая ступень диагностики при подозрении на миодистрофию Дюшенна· Исследование активности КФК сыворотки крови.
При миопатии Дюшенна происходит распад миоцитов (рабдомиолиз) и высвобождению в кровь КФК и других продуктов цитолиза. Поэтому уровень КФК крови значительно повышен. Отклонения уровня активности КФК в 100 и более раз. Таким образом, исследование активности КФК сыворотки крови может быть первый шагом в диагностике заболевания.
Показатели ЛДГ, АЛТ, АСТКроме определения КФК в биохимическом анализе крови необходимо определять активность таких ферментов как лактатдегидрогеназа (ЛДГ), и, так называемых, «печеночных ферментов» аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ). При ПМД Дюшенна активность фермента ЛДГ бывает повышена в 3-5 раз. Активность ферментов АСТ и АЛТ, имеющих внепеченочное происхождение, может быть повышена в десятки раз.
Для верификации диагноза Прогрессирующая мышечная дистрофия Дюшенна необходима генетическая диагностика.
Проведение ДНК анализаВыполнение генетического исследования начинают обычно с простого метода MLPA (Multiplex ligation-dependent probe amplification – англ., множественная лиганд-зависимая проба амплификации), позволяющего проверить наличие всех 79 экзонов в гене DMD – так может быть обнаружена делеция экзона/ов.
Выявление дупликаций проводится посредством модифицированной MLPA. Если мутация так и не была выявлена применяют секвенирование гена DMD, для выявления точечных мутаций. Различные виды генетических исследований позволяют получить более подробную информацию об изменениях структуры (мутациях) ДНК при ПМД Дюшенна. Для проведения исследования необходимо 2 мл венозной крови. Подтверждение диагноза результатами генетического исследования позволяет включить ребенка в клинические исследования, а для его родителей выработать рекомендации по пренатальной диагностике при следующих беременностях. После определения мутации (изменения гена дистрофина), матери ребенка предлагается пройти генетическое исследование для выявления носительства мутации в гене DMD. Такая информация важна для других родственников женского пола со стороны матери (ее сестер, дочерей, теток, двоюродных сестер), поскольку они также могут являться носителями данной мутации.
Возможно проведение пренатальной диагностика ПМД Дюшенна методами молекулярно-генетического исследования (материал для исследования у матери необходимо забирать до 12 недели беременности), а также предимплантационная генодиагностика в случае проведения экстракорпорального оплодотворения.
Применяемое в настоящее время противовоспалительное лечение кортикостероидами является золотым стандартом, описанным в международном руководстве по ведению пациентов с миодистрофией Дюшенна.
Данные рандомизированных контролируемых исследований показали пользу кортикостероидов для восстановления мышечной силы. Кроме основного противовоспалительного эффекта (снимает отек и воспаление, стабилизирует мышечную мембрану), терапия кортикостероидами позволяет пациенту продлить время способности к самостоятельному передвижению (ходьбы). В настоящее время прием кортикостероидов сохраняется даже после потери пациента способности ходить. Продолжение терапии поможет сдерживать развитие сколиоза, сохранять мышечную силу в верхних конечностях, поддержать дыхательную и сердечную функцию. Поскольку кортикостероиды используются пациентами с миодистрофией Дюшенна в течение длительного времени, важен мониторинг нежелательных явлений и осложнений такой терапии (например, набор веса, изменения в поведении, задержку роста и\или полового созревания и др.).
Кардиопротективная терапияКардиомиопатия и сердечная недостаточность являются основными причинами смертности у пациентов с МДД.
Целью кардиопротективной терапии является снижение нагрузки на миокард. Доказана эффективность назначения ингибиторов АПФ для профилактики дилатационной кардиомиопатии. Например, одним из первых было исследование Института миологии в Париже показавшее, что при раннем назначении ингибиторов АПФ к 15 годам кардиомиопатия сформировалась всего у 20–30 процентов пациентов вместо 70 процентов, как было раньше. Также назначаются препараты, снижающие частоту сердечных сокращений – бета-блокаторы. Это относится к стадии компенсации.
Кардиометаболическая терапия (L-карнитин, коэнзим Q10)На стадии декомпенсации специалист может рекомендовать дополнительные препараты (например, кардиотоники).
Метаболическая терапияДля профилактики остеопороза показано назначение препаратов, содержащих витамин D3 и кальций.
Генная терапияЦель этиотропного лечения устранить причину заболевания — мутацию в гене DMD, тем самым восстановить синтез дистрофина. В настоящее время в мире одобрено несколько препаратов, восстанавливающих синтез белка.
Нонсенс мутация. Стоп-кодонПриблизительно 10–15% МДД вызваны точечными мутациями, приводящими к образованию преждевременного стоп-кодона, то есть, преждевременной остановке синтеза белка дистрофина. Аталурен (Translarna, PTC Therapeutics, США) разработан для пропуска преждевременных стоп-кодонов. Аталурен связывается с рибосомной РНК и ухудшает распознавание преждевременного стоп-кодона, что позволяет восстановить трансляцию и синтез модифицированного дистрофина.
Пропуск экзона/овНарушение рамки считывания (процесса синтеза белка дистрофин) в большинстве случаев связано с делециями и дупликациями, поэтому основной разрабатываемой стратегией генотерапии являлся метод пропуска экзона или экзонов. Такой метод приводит к восстановлению рамки считывания, иными словами – возобновлению экспрессии укороченного дистрофина. Восстановление экспрессии частично функционального дистрофина должно преобразовать злокачественную форму МДД с тяжелым фенотипическим проявлением в МДБ – мышечную дистрофию Беккера. Данный метод лечения может помочь большинству пациентов с мышечной дистрофией Дюшенна. Исключение составят делеции, разрушающие актин-связывающие домены в N-концевой области или затрагивающие первый или последний экзоны и крупные хромосомные перестройки. Такие мутации достаточно редки и вместе составляют менее 10% от всех описанных мутаций в гене DMD. В настоящее время одобрены три препарата, использующие метод пропуска экзона с помощью антисмысловых олигонуклеотидов АОН.
Ведутся и другие разработки препаратов генной терапииСамым ожидаемым является универсальный препарат или метод, который сможет применяться при любом виде мутации в гене дистрофин.
Таким препаратом можно назвать мини и микродистрофин или метод CRISPR/Cas9.
Внедрение мультидисциплинарного подхода и соблюдение стандартов оказания медицинской помощи позволило увеличить продолжительность жизни пациентов с мышечной дистрофией Дюшенна в мире по данным ретроспективного анализа в среднем до 27,9 лет (от 23 до 38,6 лет).
Ссылки на использованные статьи:Saito T, Kawai M, Kimura E et al. Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul Disord. 2017 Feb;27(2):107-114. doi: 10.1016/j.nmd.2016.11.012.
Passamano L., Taglia A., Palladino A. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012 Oct; 31(2): 121–125.
Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard JP, Roy R, et al. Dystrophin expression in muscles of Duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65(4):371–86. https://doi.org/10.1097/01. jnen.0000218443.45782
Статья носит информационный, ознакомительный характер. Диагностика заболевания и лечение возможны только под наблюдением специалистов
Поделиться статьей:
Новый препарат, разработанный под руководством профессора медицинской генетики Тосифуми Йокоты из Университета Альберты, может быть полезен 45 % пациентов с мышечной дистрофией Дюшенна — это значительно больше, чем доступные в настоящее время методы лечения.
Миодистрофия Дюшенна поражает одного из 3500–5000 новорожденных мальчиков и одну из 50 000 девочек. Эта болезнь вызвана мутациями гена белка дистрофина, расположенного на Х-хромосоме: поскольку она у мужчин одна, сильный пол в данной ситуации оказался более уязвим. Дистрофин соединяет цитоскелет мышечной клетки с окружающей ее тканью — только так мышца может нормально работать. Ген, ответственный за синтез дистрофина, самый большой в человеческом геноме. Он состоит из 79 отдельных секций (экзонов), если хотя бы одной из них не хватает, организм не может вырабатывать дистрофин.
У больных миодистрофией Дюшенна белок дистрофин полностью или почти полностью отсутствует. Мышцы постепенно ослабевают, и к подростковому возрасту ребенок становится тяжелым инвалидом, прикованным к коляске и дыхательному аппарату. Рано или поздно патология также затрагивает сердце. При этом больные чаще всего умирают от дыхательной недостаточности в возрасте 20–25 лет.
Новый класс лекарств — ДНК-подобные молекулы, называемые антисмысловыми олигонуклеотидами, — использует так называемый «пропуск экзонов», который позволяет организму пропустить поврежденные части генетической инструкции и произвести необходимый белок для восстановления мышечной ткани, пусть не полностью аналогичный природному дистрофину, но все-таки способный выполнять его функции. Миодистрофия при применении таких препаратов переходит в менее тяжелую форму. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило четыре антисмысловых олигонуклеотида для лечения миодистрофии Дюшенна. Среди них есть вилтоларсен, созданный на основе предыдущих исследований Тосифуми Йокоты, но каждый из этих олигонуклеотидов имеет ограниченное применение, воздействуя только на один конкретный участок гена.
Теперь группа исследователей в области медицинской генетики Университета Альберты объединила шесть таких молекул, чтобы создать «коктейль», который может помочь большему числу пациентов. «Каждая из ранее разработанных пропускающих экзон молекул была способна лечить только около 10 % пациентов с МДД, потому что они имеют разные мутации своих экзонов в разных местах внутри гена, — говорит профессор Йокота. — Наш подход заключается в одновременном пропуске более 11 экзонов, что позволит нам лечить примерно 45 % пациентов».
Команда Йокоты протестировала новый синтетический препарат на образцах мышечной ткани пациентов и на мышах. Они обнаружили признаки выработки дистрофина, наращивания мышечной массы и улучшения работы сердца. Следующим шагом будет проведение токсикологических испытаний и выполнение нормативных требований для проведения клинических испытаний.
Исследование опубликовано в журнале Proceedings of the National Academy of Sciences.
Мышечная дистрофия (миодистрофия) Дюшенна — это редкое врожденное заболевание, которое бывает только у мальчиков. Оно проявляется после семи лет — ребенок начинает падать без видимых причин. Постепенно мышечная слабость нарастает, в последнюю очередь перестают работать дыхательные мышцы и сердце. Лечения от этого заболевания не существует, и обычно человек с таким диагнозом не доживает до 20-25 лет.
История Кирилла
Узнав об этой истории благодаря многочисленным сообщениям болельщиков, сине-бело-голубые связались с фондом «МойМио» и пригласили семью Шушкиных на матч. Врачи не рекомендуют мальчику передвигаться на дальние расстояния, поэтому Кирилл и его мама приехали на игру «Зенита» в Москве.
Главным сюрпризом для Кирилла стала встреча с любимым игроком. В день матча действует особый режим для футболистов: они полностью концентрируются на игре. Но ради знакомства с Кириллом было сделано исключение.
После утренней разминки в лобби отеля, где жили Шушкины, появился Артем Дзюба.
— Спасибо тебе большое, что приехал, — сказал нападающий Кириллу. — Мне со всего Инстаграма столько людей о тебе писали! Спасибо тебе, что переживаешь и болеешь за нас, это очень приятно. Это дорогого стоит, спасибо тебе!
Нападающий подарил мальчику футболку с автографом и билет на первый матч в жизни Кирилла.
— Дзюба такой же, как и на фотках, — поделился впечатлениями Кирилл. — Только похудел немного.
Вечером Кирилл Шушкин впервые побывал на стадионе, первый раз увидел вживую матч «Зенита» и игру Артема Дзюбы. Нападающий забил гол и отдал результативную передачу, чем порадовал 15 миллионов болельщиков сине-бело-голубых, но самым счастливым из них в этот день стал Кирилл.
Футбольный клуб «Зенит» выражает благодарность за содействие в исполнении мечты изданию «Такие дела» и фонду «МойМио».
Врач-невролог ФГАУ «НМИЦ здоровья детей» Минздрава России, кандидат медицинских наук Подклетнова Татьяна Владимировна провела в Республике Калмыкия первую внутривенную инфузию препарата «Виондис 53» пациенту с диагнозом мышечная дистрофия Дюшенна.
Миодистрофия Дюшенна — наследственное заболевание, которое характеризуется ранним началом, прогрессирующим течением в виде стремительной атрофии мышц, в сочетании с сердечно-сосудистыми, дыхательными костно-суставными и когнитивными нарушениями.
Детям с таким диагнозом сложно бегать, прыгать, подниматься по ступенькам и вставать с пола. Они часто падают и получают переломы рук или ног. Мышечная слабость у них быстро и неуклонно прогрессирует, и без лечения это приводит к потере способности ходить. Также возникают проблемы с дыханием и работой сердца. Не получая адекватной терапии большинство детей могут передвигаться только с помощью инвалидной коляски уже в возрасте 10-12 лет и умирают от респираторных осложнений к 20 годам.
«Виондис 53» (голодирсен, Vyondys 53, Sarepta Therapeutics) — новый препарат, предназначенный для лечения миодистрофии Дюшенна у пациентов с подтвержденной мутацией гена дистрофина, подходящей для терапевтического пропуска экзона 53. Он обеспечивает значимый рост уровня белка дистрофина в скелетных мышцах у части больных миодистрофией Дюшенна, что приводит к замедлению прогрессирования двигательных нарушений, стабилизации состояния дыхательной системы.
Терапия «Виондис 53» должна осуществляться постоянно, препарат необходимо вводить внутривенно раз в неделю.
Первая инфузия данного препарата, который был закуплен региональным Министерством здравоохранения, прошла успешно! Мальчик после введения чувствует себя хорошо.
Татьяна Владимировна Подклетнова обучила коллег-медиков региона правильной технике введения препарата и в дальнейшем специалисты смогут проводить данную манипуляцию самостоятельно.
Поздравляем отечественное медицинское сообщество со столь значимым событием в лечении миодистрофии Дюшенна в нашей стране. Благодарим Татьяну Владимировну за чуткое руководство первой инфузии уникального препарата.
«Хотим мечтать дольше» — фильм, в котором главные герои — мальчики, подопечные фонда «Родительский проект мышечной дистрофии», ежедневно борющиеся с мышечной дистрофией Дюшенна, приглашают зрителей в свой мир. Они рассказывают о своих увлечениях, мечтах и повседневной жизни.
Мышечная дистрофия Дюшенна начинается с, казалось бы, незначительных проблем с ходьбой.Однако со временем болезнь прогрессирует. В конце концов больной мальчик перестает ходить и начинает пользоваться инвалидной коляской, затем респиратором и энтеральным питанием. При этом заболевании мышцы деградируют, и в конце концов каждое малейшее движение становится для больного испытанием.
Мышечная дистрофия Дюшенна является генетической. В результате генной мутации у больного исчезает дистрофин, т.е. белок, отвечающий за правильное функционирование мышц.
Мышечная дистрофия Дюшенна считается одним из наиболее распространенных и быстро прогрессирующих нервно-мышечных заболеваний.По разным данным, им страдает один из примерно 3-3,5 тысяч человек. мальчики. Это необратимое и прогрессирующее заболевание, обычно приводящее к атрофии мышц относительно быстро. Его развитие тесно связано с полом ребенка — за исключением нескольких случаев — ему подвержены мальчики. Вылечить на данном этапе невозможно. Поэтому лечение направлено на замедление прогрессирования заболевания. Вот почему так важно, чтобы пациенты имели доступ к систематической физической реабилитации и современным методам лечения.
Каково жить с болезнью? как прогрессируют симптомы? Посмотрите видео, показывающее повседневную жизнь мальчиков, страдающих мышечной дистрофией Дюшенна (прогрессирующая мышечная атрофия). Они рассказывают о своих мечтах, заботах, буднях, отмеченных борьбой с болезнью. В фильме «Мы хотим дольше мечтать» снялись воспитанники Фонда Родительского Проекта Мышечной Дистрофии: Тимек, Себастьян, Филип, Миколай, Михал, Аркадиуш, Патрик.
Спасибо, что дочитали нашу статью до конца. Если это вас интересует, будьте в курсе и присоединяйтесь к группе подписчиков наших социальных профилей. Подпишитесь на Facebook, Подпишитесь на Instagram.
.90 000 причин, симптомов, диагностики и лечения 90 001Врожденная мышечная слабость, развивающаяся по мере развития организма, известна в медицине как «мышечная дистрофия Дюшенна». Это заболевание встречается только у мужчин. Понимание природы патологии помогает облегчить ее течение и помочь детям справиться с начальными симптомами.
Мышечная дистрофия Дюшенна — генетическая патология, обусловленная нарушением строения мышечных волокон.Они постепенно распадаются, и человек теряет способность двигаться. Заболевание проявляется уже в младенчестве. Сначала возникают мышечные нарушения, затем сочетаются деформации скелета. Клиническая картина дополняется гормональными и психическими нарушениями. 
Впервые в 1861 году описал миодистрофию французский невропатолог, имя которого впоследствии было названо. Часто диагностируется: 1 из 3500 новорожденных. Радикального лечения нет. Предлагаемая врачами терапия носит чисто симптоматический характер.Пациенты с таким диагнозом редко переживают рубеж в 30 лет.
Мышечная дистрофия является следствием отклонения в генетическом коде ДНК. Мутация происходит в гене, расположенном на Х-хромосоме, один из ее участков отвечает за выработку специфического белка – дистрофина. Вещество на микроскопическом уровне составляет основу мышечных волокон и выполняет несколько функций:
При этом заболевании дистрофин отсутствует или плохо синтезируется. Уровень «нормального» белка составляет менее 3%. Эта мутация разрушает волокна в мышцах. Они постепенно регенерируют, замещаясь жировой тканью и соединительной тканью. В результате человек теряет способность двигаться.
Что такое наследственность мышечной дистрофии Дюшенна? Заболевание передается по рецессивному признаку. У человека все пары парные. Чтобы наследственное заболевание выглядело патологическим, генетический дефект должен возникнуть в одной хромосоме или в сходных частях обеих.Во втором случае это рецессивный тип наследования.
Если генетический дефект диагностирован только на одной хромосоме, но болезнь прогрессирует, то это является преобладающим признаком передачи инфекции. Возможен рецессивный тип с одновременным изменением идентичных структур ДНК. Когда вторая хромосома полностью здорова, патология не развивается. Поэтому дистрофия диагностируется только у мужчин. У них есть одна Х-хромосома в их генетическом составе, а другая (U) - пара.
Что наука говорит о прекрасном поле? Мышечная дистрофия Дюшенна у девочек диагностируется редко.Для этого две патологические Х-хромосомы должны совпадать по генотипу, что маловероятно. Девочки могут выступать только переносчиками болезни и передавать ее своим сыновьям. 
Заболевание наложило отпечаток на нервно-мышечную систему. Его симптомы можно наблюдать у детей 2-3 лет. Родители начинают замечать, что ребенок отстает в физическом развитии от сверстников. Патологический процесс быстро прогрессирует и распространяется на нижние конечности.Затем он переходит на другие части мышц.
Повреждения мышечного аппарата и чрезмерные нагрузки приводят к искривлению конечностей. У больных детей также отмечаются изменения в работе сердца, умственная отсталость.
Все патологические симптомы заболевания можно разделить на несколько групп:
Рассмотрим клинические особенности каждой группы более подробно.
Дети рождаются без серьезных проблем со здоровьем. Однако через несколько месяцев их двигательное развитие начинает задерживаться. Такие дети менее активны. Врачи и родители до сих пор не замечают явных отклонений, списывая все на особенности темперамента.
Начальные симптомы заболевания появляются после первых шагов. Дети с дистрофией Дюшенна продолжают падать и двигаться на носочках.Если большинство их сверстников крепко стоят на ногах, то они упорно изо всех сил пытаются двигаться.
Следующей стадией проявления болезни является период, в котором дети приобретают способность говорить. Они начинают жаловаться родителям на слабость и быструю утомляемость. Прыжки на детской площадке, бег, лазание по турникам – все эти виды деятельности не приносят им удовольствия.
Что еще имеет симптомы мышечной дистрофии Дюшенна? Нарушение Говерса считается своеобразным проявлением болезни.Каждый раз, когда вы пытаетесь встать с пола, ваш ребенок использует руки, чтобы помочь слабым мышцам ног. Для этого он опирается на конечности, перекручивая их по всему телу.
Постепенное прогрессирование заболевания приводит к тому, что к 10-12 годам больные дети теряют способность самостоятельно передвигаться. Большинству из них нужна инвалидная коляска. Способность удерживать тело в вертикальном положении сохраняется только до 16 лет. 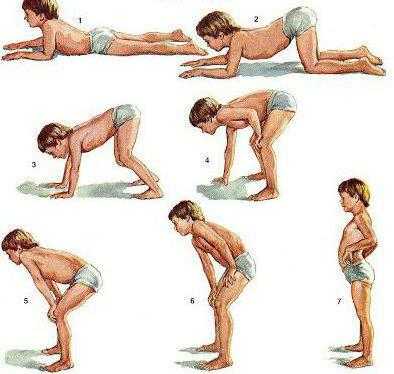
К этой группе относятся симптомы, связанные с мышечными расстройствами.Мышечная дистрофия Дюшенна проявляется повышенным поясничным сгибанием, которое дополняется искривлением грудного отдела позвоночника и давлением. У многих детей меняется форма стопы. Остеопороз развивается со временем. Эти симптомы еще больше углубляют клиническую картину и способствуют усугублению двигательных нарушений.
Прогрессирующая мышечная дистрофия всегда сопровождается изменениями. сердечная мышца. Как правило, развивается кардиомиопатия. Клинически проявляется перепадами давления и нарушениями сердечного ритма.Границы основных мышц тела увеличиваются, но при этом его функциональные возможности значительно снижаются. В результате возникает сердечная недостаточность.
Сочетание этих пороков с нарушением функции дыхания часто приводит к летальному исходу.
Этот симптом считается необязательным, но возможным. Его появление может быть связано с дефицитом одного из видов дистрофина – аподистрофина, содержащегося в головном мозге. При этом слабость мышц и тяжесть психических расстройств не связаны друг с другом.Невозможность посещать детский сад и школу только усугубляет когнитивные нарушения. 
Различные виды эндокринных нарушений диагностируются у 30-50% больных. Они могут выражаться в виде ожирения или недоразвития половых органов. Избыточные отложения чаще всего наблюдаются в области груди, плечевого пояса и ягодиц. Больные дистрофией Дюшенна склонны к низкорослости.
Диагностика миодистрофии Дюшенна основывается на нескольких видах тестов, основным из которых является тест ДНК.Обнаружение дефекта Х-хромосомы в месте, отвечающем за синтез дистрофина, считается окончательным подтверждением диагноза.
Среди других методов диагностики, применяемых сегодня:
Прогрессирующая мышечная дистрофия у ребенка означает наличие патологической Х-хромосомы в генотипе матери. Только в отдельных случаях женщина полностью здорова. Наличие дефектного гена представляет угрозу для будущих беременностей. Поэтому таким семьям рекомендуется посещать генетика.
При наступлении второй беременности супружеская пара проходит так называемую пренатальную диагностику.Он включает в себя изучение генотипа ребенка еще в утробе матери. Такое исследование исключает риск наследственных заболеваний, в том числе мышечной дистрофии Дюшенна.
Пренатальная диагностика основана на использовании клеточного материала. Его получают с помощью различных процедур: биопсии ворсин хориона, амниоцентеза и др. Вышеуказанные манипуляции сопряжены с определенным риском для плода, но с их помощью можно со 100% гарантией диагностировать генетическое заболевание. 
Мышечная дистрофия Дюшенна — неизлечимое заболевание.Но больные с таким диагнозом не должны оставаться прикованными к постели. Чтобы помочь ребенку продлить период физической активности, современная медицина предлагает несколько способов.
С этой целью больным назначают стероиды и бета-адреномиметики. Применение последних («Альбутерол», «Формотерол») ненадежно. Поэтому говорить об их эффективности сегодня не приходится. Такие препараты используются только в качестве экспериментального лечения.
Основу терапии составляют стероиды.Регулярное их использование позволяет на некоторое время восполнить силы мышц. Врачи предполагают, что такие лекарства могут замедлить прогрессирование заболевания, а также предотвратить возникновение сколиоза. Однако возможности стероидов ограничены. Мышечная дистрофия Дюшенна в любом случае будет продолжать развиваться.
Дополнительно назначают больным на сердце. В основном это ингибиторы АТФ, антиаритмические и метаболические препараты. Они позволяют противостоять кардиологическим аспектам заболевания.
Немедикаментозная терапия включает назначение физиотерапевтической и ортопедической помощи. В первом случае речь идет о различных техниках массажа и плавания. Физиотерапевтический эффект помогает надолго сохранить подвижность и гибкость суставов. Умеренная активность положительно влияет на течение болезни. С другой стороны, малоподвижность и постельный режим могут только ухудшить клиническую картину. Поэтому врачи рекомендуют оставаться физически активными как можно дольше.
Ортопедическая помощь является важным компонентом лечения пациентов с мышечной дистрофией Дюшенна. Лечение лекарственными препаратами в сочетании со специальными аппаратами позволяет значительно упростить им жизнь. Их перечень весьма разнообразен: различные вертикальные стойки, приспособления для самопомощи с удобным положением, инвалидные коляски с электроприводом, корсеты для позвоночника, шины для ног и многое другое. 
Мышечная дистрофия Дюшенна — тяжелое генетическое заболевание, первые признаки которого обнаруживают у малышей первых месяцев жизни.Мальчикам сначала тяжело ходить, а потом они не могут просто оторваться от пола. Медикаментозное лечение стероидами существенно меняет течение патологического процесса. Препараты помогают восстановить мышечную силу с течением времени.
Достаточно сложно предсказать, когда именно пациент начнет пользоваться инвалидной коляской. Обычно спрос на это устройство появляется в возрасте 8-11 лет. При дальнейшем развитии мышечной слабости больному трудно удерживать положение тела, могут возникнуть осложнения.
Какой диагноз поставили врачи при диагностике мышечной дистрофии Дюшенна? Заболевание может значительно сократить продолжительность жизни. Однако сегодня большинство юношей достигают совершеннолетия при условии качественного медикаментозного и физиотерапевтического лечения.
.90 000 Мышечная дистрофия — приговор или испытание на прочность? Мышечная дистрофия у детей: симптомы, лечение, диагностика, причины.Х-сцепленная наследственная патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстрым распространением и усилением мышечной слабости. Вначале поражаются мышцы тазового пояса и бедер, затем плечи и спина, постепенно обездвиживание. Миодистрофия сопровождается деформациями скелета и поражением сердца.Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, анализ ДНК, биопсию мышц. Лечение симптоматическое. В связи с ослаблением дыхательной мускулатуры в конечной стадии заболевания необходима искусственная вентиляция легких.
Профилактические мероприятия предназначены для выявления женщин-носителей аномального гена дистрофина и предупреждения рождения больного ребенка. В рамках профилактических мероприятий проводятся генетические консультации для пар, планирующих беременность, консультации для беременных и пренатальная ДНК-диагностика.
Мышечная дистрофия, или, как ее еще называют врачи, миопатия, является генетическим заболеванием. В редких случаях развивается от внешних причин. Чаще всего это наследственное заболевание, характеризующееся мышечной слабостью, дегенерацией мышц, уменьшением диаметра волокон скелетных мышц, а в особо тяжелых случаях и мышечных волокон внутренних органов.
При этом заболевании мышцы постепенно теряют способность сокращаться.Происходит постепенный распад. Мышечная ткань медленно, но верно замещается жировой тканью и соединительными клетками.
Прогрессирующая стадия характеризуется следующими признаками:
Медицина до сих пор не может назвать все механизмы, запускающие мышечную дистрофию. Одно можно сказать с абсолютной уверенностью: все причины кроются в изменении в нашем организме набора доминантных хромосом, отвечающих за метаболизм белков и аминокислот. Без правильного усвоения белка не будет нормального роста и функции мышечной и костной ткани.
Течение заболевания и его форма зависят от типа мутировавших хромосом:
Диагностические мероприятия разнообразны.Есть много состояний, которые так или иначе напоминают непрямую миопатию. Наследственность является наиболее частой причиной мышечной дистрофии. Лечение возможно, но оно будет долгим и трудным. Не забудьте собрать информацию о распорядке дня и образе жизни пациента. Как он питается, ест ли мясо и молочные продукты, употребляет ли алкогольные напитки или наркотики. Эта информация особенно важна при диагностике мышечной дистрофии у подростков.
Данные необходимы для составления плана проведения диагностических мероприятий:
Изучая развитие прогрессирующей мышечной дистрофии на протяжении веков, врачи выделили следующие виды заболевания:
Это наиболее распространенные формы заболевания. Некоторые из них уже можно успешно преодолевать благодаря развитию современной медицины.Некоторые имеют наследственные причины, хромосомные мутации и не поддаются терапии.
Инвалидность является результатом появления и прогрессирования миопатий различного генеза и этиологии. Сильная деформация скелетных мышц и позвоночника приводит к частичной или полной потере подвижности.

Прогрессирующая мышечная дистрофия часто приводит к развитию почечной, сердечной и дыхательной недостаточности.У детей - к задержке умственного и физического развития. У подростков - при нарушении интеллектуальных и умственных способностей, задержке роста, карликовости, ухудшении памяти и потере способности к обучению.
Это одна из самых тяжелых форм. К сожалению, современная медицина не смогла помочь пациентам с прогрессирующей мышечной дистрофией Дюшенна приспособиться к жизни. Большинство больных с этим диагнозом с детства являются инвалидами и не живут более тридцати лет.
Клинически проявляется в возрасте двух-трех лет. Детям нельзя играть на свежем воздухе со сверстниками, они быстро устают. Часто наблюдается задержка в развитии речи и познания. К пяти годам мышечная слабость и недоразвитие скелета у ребенка становятся совершенно очевидными. Походка выглядит странно - слабые мышцы ног не позволяют больному ходить плавно, не шатаясь из стороны в сторону.
Родители должны бить тревогу как можно скорее. Как можно скорее выполните серию генетических тестов, чтобы поставить точный диагноз.Современные методы лечения помогут больному вести приемлемый образ жизни, хотя и не восстановят полностью рост и функцию мышечной ткани.
Эта форма мышечной дистрофии была исследована Беккером и Кинером еще в 1955 году. Это известно в медицинском мире как мышечная дистрофия Беккера или Беккера-Кенера.

Основные симптомы такие же, как при болезни Дюшенна. Причины развития также кроются в нарушении генного кода.Но в отличие от дистрофии Дюшенна форма болезни Беккера протекает легко. Больные данным типом заболевания могут вести практически полноценную жизнь и дожить до преклонного возраста. Чем раньше будет диагностировано заболевание и начато лечение, тем больше шансов у больного вести нормальную человеческую жизнь.
Отсутствует замедление развития психических функций человека, характерное для злокачественной мышечной дистрофии Дюшенна. При рассматриваемом заболевании кардиомиопатия и другие нарушения в работе сердечно-сосудистой системы встречаются очень редко.
Эта форма заболевания протекает довольно медленно и протекает в легкой форме. Чаще всего первые симптомы заболевания заметны в возрасте шести-семи лет. Но иногда (около 15% случаев) заболевание проявляется только через тридцать-сорок лет. В ряде случаев (10%) ген дистрофии вообще не просыпается на протяжении всей жизни больного.
Как следует из названия, это относится к мышцам лица, плечевого пояса и верхних конечностей.Отставание лопатки от спины и неравномерное положение уровня плеч, искривление плечевой дуги - все это свидетельствует о слабости или полной дисфункции передней зубчатой, трапециевидной и со временем двуглавой, задней дельтовидной мышц в процессе.
У опытного врача при взгляде на пациента может сложиться ложное впечатление, что у него экзофтальм. При этом функция щитовидной железы остается нормальной, обмен веществ чаще всего не изменяется. Как правило, сохраняются и интеллектуальные способности больного.У пациента есть все возможности вести полноценный, здоровый образ жизни. Современные лекарства и лечебная физкультура помогут визуально облегчить симптомы лицевой мышечной дистрофии.
Наследуется по аутосомно-доминантному типу в 90% случаев. Поражает мышечную и костную ткани. Миотоническая дистрофия является очень редким явлением, с частотой 1 на 10 000, но эта статистика занижена, поскольку эта форма заболевания часто не диагностируется.
Дети, рожденные от матерей с миотонической дистрофией, часто страдают так называемой врожденной миотонической дистрофией. Проявляется ослаблением мимических мышц. Параллельно нередко наблюдается дыхательная недостаточность новорожденных, перебои в работе сердечно-сосудистой системы. Нередко можно заметить задержку психического развития, задержку психоречевого развития у молодых больных.
В классических случаях гипотензия заметна с младенческого возраста.Характеризуется уменьшением объема мышечной и костной ткани наряду с контрактурами в суставах рук и ног. В анализах повышается активность КФК в сыворотке. Биопсия пораженных мышц выявляет стандартную картину мышечной дистрофии.

Данная форма непрогрессирующая, интеллект больного практически всегда сохранен. К сожалению, многие пациенты с врожденной мышечной дистрофией не могут самостоятельно передвигаться. Позже может развиться дыхательная недостаточность.Компьютерная томография иногда выявляет гипомиелинизацию слоев белого вещества головного мозга. Это не имеет известных клинических симптомов и чаще всего не влияет на психическую адекватность и жизнеспособность пациента.
Отказ многих подростков от еды влечет за собой необратимую дисфункцию мышечной ткани. Если в течение сорока дней в организм не поступают аминокислоты, процессы синтеза белковых соединений не происходят – мышечная ткань погибает на 87%.Поэтому родители должны следить за питанием своих детей, чтобы они не сидели на новомодных аноректических диетах. Рацион подростка должен ежедневно включать мясо, молочные продукты и растительные источники белка.
При запущенных расстройствах пищевого поведения может наблюдаться полная атрофия отдельных участков мышц, часто как осложнение появляется почечная недостаточность, сначала острая, а затем хроническая.
Дистрофия — тяжелое хроническое наследственное заболевание.Полностью вылечить его невозможно, но современная медицина и фармакология позволяют корректировать симптомы заболевания, чтобы жизнь больных была максимально комфортной.
Перечень препаратов, необходимых больным для лечения мышечной дистрофии:
Мышечная дистрофия представляет собой группу наследственных заболеваний, характеризующихся прогрессирующей симметричной атрофией скелетных мышц без болей или потери чувствительности в конечностях. Как это ни парадоксально, пораженные мышцы могут увеличиваться за счет разрастания соединительной ткани и жировых отложений, что создает ложное впечатление сильных мышц.
На сегодняшний день лекарства от мышечной дистрофии не существует. Различают четыре основных вида данной патологии. Наиболее часто встречается мышечная дистрофия Дюшенна (50% всех случаев). Заболевание обычно начинается в раннем детстве и заканчивается смертельным исходом в возрасте 20 лет. Мышечная дистрофия Беккера развивается медленнее, больные живут более 40 лет. Лопаточно-лицевая и лимо-поясная дистрофии обычно не влияют на продолжительность жизни.
развитие мышечной дистрофии.из-за разных генов. Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера вызываются генами, обнаруженными в половой хромосоме, и поражают только мужчин. Плечево-лопаточно-лицевые и конечностно-поясничные дистрофии не связаны с половыми хромосомами; они затрагивают как мужчин, так и женщин.
Все типы мышечной дистрофии вызывают прогрессирующую атрофию мышц.
Врач осматривает ребенка, задает вопросы о болезнях членов семьи и назначает определенные анализы.Если родственник страдал мышечной дистрофией, врач узнает об их дистрофии. Анализируя полученные данные, можно спрогнозировать, чего ожидает ребенок. Если в семье не было больных мышечной дистрофией, электромиография позволит оценить работу нервов в пораженных мышцах и определить наличие мышечной дистрофии; исследование кусочка мышечной ткани () может выявить клеточные изменения и наличие жировой ткани.
В медицинских учреждениях, оснащенных самым современным оборудованием для молекулярно-биологических и иммунологических исследований, можно точно определить, будет ли ребенок страдать мышечной дистрофией.Эти центры также могут проверить родителей и родственников на наличие генов, определяющих развитие мышечной дистрофии Дюшенна и мышечной дистрофии Беккера.
В зависимости от тяжести заболевания и времени его начала различают:
Дистония Дюшенна проявляется в раннем возрасте (от 3 до 5 лет). Больные дети шаркают, с трудом поднимаются по лестнице, часто падают и не могут бежать. Когда они поднимают руки, их лопатки «отстают» от туловища — симптом, называемый «лопастями крыльев».Обычно ребенок с мышечной дистрофией прикован к инвалидной коляске к 9-12 годам. Прогрессирующая слабость сердечной мышцы приводит к смерти от внезапной сердечной недостаточности, дыхательной недостаточности или инфекции.
Хотя дистрофия Беккера имеет много общего с дистрофией Дюшенна, она прогрессирует гораздо медленнее. Симптомы начинаются примерно в 5 лет, но после 15 лет больные дети обычно еще способны ходить, а иногда и значительно позже.
Плечево-лопаточно-лицевая дистрофия развивается медленно и протекает относительно легко.Чаще всего она начинается в возрасте до 10 лет, но может проявиться и в раннем подростковом возрасте. Младенцы, у которых впоследствии возникает эта патология, в младенчестве плохо сосут грудь; когда они становятся старше, они не сжимают губы, как в свист, а поднимают руки над головой. У больных детей лица отличаются малоподвижностью при смехе или плаче, иногда отмечается отличное от нормального выражение лица.
Если вы обеспокоены тем, что у вашего ребенка может развиться мышечная дистрофия, вы можете принести фотографии или видео, иллюстрирующие ваши конкретные опасения.Возьмите с собой родственника или друга, который также выслушает информацию от вашего врача.
На сегодняшний день не существует лекарства, способного остановить прогрессирование мышечной атрофии при мышечной дистрофии. Тем не менее, ортопедические устройства, а также лечебная физкультура, физиотерапия и операции по исправлению контрактур могут на некоторое время заставить ребенка или подростка двигаться.
Члены семьи с мышечной дистрофией в анамнезе должны обратиться за медицинской консультацией по вопросам генетики, чтобы определить, существует ли риск передачи заболевания будущему ребенку.
Некоторые виды мышечной дистрофии сокращают продолжительность жизни, часто поражая мышцы, связанные с дыханием. Даже при улучшенном механическом дыхании люди с мышечной дистрофией Дюшенна — наиболее распространенным типом мышечной дистрофии — умирают от дыхательной недостаточности в возрасте до 40 лет.
Многие виды мышечной дистрофии также могут снижать работоспособность миокарда. Проблемы с приемом пищи могут возникнуть, если глотательные мышцы заняты.
Когда мышцы ослабевают, подвижность становится проблемой. Многим людям, у которых развивается мышечная дистрофия, может понадобиться инвалидное кресло. Однако длительная иммобилизация суставов, связанная с использованием инвалидной коляски, может усугубить контрактуру, при которой конечности вращаются и блокируются во внутреннем положении.
Контрактуры также могут играть роль в развитии сколиоза, вызывая искривление позвоночника, что еще больше снижает функцию легких у людей с мышечной дистрофией.
Поскольку респираторные инфекции могут стать проблемой на более поздних стадиях мышечной дистрофии, важно делать прививки от пневмонии, а также регулярно делать прививки от гриппа.
Мышечная дистрофия (миопатия) — генетическое патологическое хроническое наследственное нервно-мышечное заболевание, характеризующееся прогрессирующей мышечной слабостью и дегенерацией (часто уменьшением диаметра (толщины) волокон скелетных мышц, а в ряде случаев и мышечных волокон внутренних органов).В процессе мышечной дистрофии пораженные мышцы систематически теряют способность сокращаться и распадаться, уступая место соединительной и жировой ткани.
Результатом появления и прогрессирования данного вида заболевания является инвалидность, которая проявляется значительной деформацией позвоночника и потерей его двигательных навыков. Этот вид заболевания поражает и другие системы организма – приводит к развитию сердечной и дыхательной недостаточности, нарушению интеллектуальных и умственных способностей, ухудшению памяти и потере обучаемости.
К формам мышечной дистрофии относятся:
Причины мышечной дистрофии до конца не выяснены.Среди основных причин они называют мутацию гена, отвечающего за способность мышечных клеток вырабатывать особые белки.
На сегодняшний день ученым удалось обнаружить ген, вызывающий патологию Дюшенна. Он расположен на хромосоме Х. Дефектный ген передается от матери к сыну, у которого заболевание развивается в возрасте от двух до пяти лет.
Причинами развития данного вида заболевания у женщин являются изменения гормонального фона, появляющиеся в связи с наступлением беременности, началом менструального цикла, климактерическим периодом.
Первые симптомы этого вида заболевания появляются с раннего детства (около 2 лет). Первые симптомы заключаются в том, что ребенок часто падает, довольно быстро утомляется даже при минимальных физических нагрузках, неуклюж в движениях. Первые симптомы особенно заметны на фоне сверстников и сверстников.
Проявление мышечной дистрофии у детей первого года жизни сопровождается появлением резкого снижения развития по сравнению с исходной жизнедеятельностью и ростом.Ребенок утрачивает приобретенные ранее навыки – теряет способность самостоятельно держать головку и переворачиваться. Часто продолжительность жизни таких детей не превышает трех лет.
Развитие данного заболевания у взрослых сопровождается снижением мышечного тонуса, что приводит к нарушению походки, атрофии скелетных мышц, постоянному чувству усталости и отсутствию мышечных болей. Наряду с этим мышечная дистрофия приводит к разрастанию и разрастанию икроножных мышц.

В процессе диагностики врач собирает семейный анамнез, который включает в себя выявление наличия данного вида патологии у родных и близких, выявление характера дистрофии.Сбор данных способствует разработке дальнейшего прогноза для пациента.
Ваш врач назначает электромиографию, которая проверяет электрическую активность мышечной ткани (выявляет первичную мышечную дистрофию, берет образец ткани для исследования на наличие дегенеративных изменений в тканях и жировых отложений).
Современное оборудование позволяет проводить иммунологический и молекулярно-биологический анализы, помогающие определить вероятность развития данного вида заболевания у детей.
Мышечная дистрофия – неизлечимое заболевание.Все лечебные мероприятия должны быть направлены на торможение деструктивных процессов, происходящих в мышечной системе человека. Для этого больному назначают прием витаминов группы В, кортикостероидов и аденозинтрифосфатов.
Препараты на основе фетальных стволовых клеток способствуют торможению (незначительному купированию) дегенеративного процесса.
Эффективным методом лечения мышечной дистрофии является лечебная физкультура (комплекс специализированных физических упражнений), позволяющая минимизировать развитие деформаций и контрактур.
К эффективным методам относится использование лечебного массажа.
При опасности поражения мышц органов дыхания выполняются дыхательные упражнения.
Тестирование женщины на патологические гены (особенно если заболевание есть у родственницы) является неотъемлемой частью процесса планирования беременности. Выявление дефектного гена возможно даже у плода во время его внутриутробного развития. Для этого исследуют анатомическую жидкость, клетки плода и кровь.
Мышечная дистрофия представляет собой ряд заболеваний, характеризующихся истощением или ослаблением различных мышц, наиболее распространенными из которых являются наследственные заболевания.
Существует несколько форм этого заболевания, различающихся по следующим характеристикам: расположение мышц, степень атрофии или слабости, возраст начала, тип приобретения, скорость прогрессирования.
Причины заболевания точно не известны, считается, что это наследственное заболевание, связанное с нарушением обмена веществ в мышцах.Некоторые формы развиваются только у мальчиков.
Симптомы мышечной дистрофии
Основным симптомом является прогрессирующая мышечная слабость.
Дистрофия Дюшенна. Чаще развивается у мальчиков и выявляется, когда ребенок пытается ходить самостоятельно. Симптомы этого вида мышечной дистрофии следующие:
затруднение вставания из положения сидя или лежа;
Частые падения ребенка;
- походка "ладан";
Затрудненный бег и прыжки;
трудности в обучении;
Увеличение икроножных мышц.
Дистрофия Беккера. Симптомы этого типа мышечной дистрофии аналогичны симптомам дистрофии Дюшенна, за исключением того, что они мягче и развиваются медленнее. Первые симптомы наблюдаются в подростковом возрасте и даже после 20 лет.
Миотоническая дистрофия (болезнь Штейнерта). Благодаря ему невозможно расслабить мышцы, когда захочется. В первую очередь это касается лицевых мышц. Начинается после полового созревания.
Дистрофия лопатки. Внешний вид человека характерен: лопатки выступают, как крылья, когда человек поднимает руки или плечи.Развивается у подростков.
Тазовая дистрофия. Страдают мышцы плеч и бедер, отмечается невозможность поднять стопу. Развивается в раннем детстве и прогрессирует.
врожденная дистрофия. Развивается сразу после рождения или появляется до 2 лет. Различают легкие формы и тяжелое течение.
Глазная дистрофия. Первый признак – опущенные веки. Отмечается слабость мышц глаз, шеи, лица, затруднение глотания.Эта форма проявляется в зрелом возрасте (40-50 лет).
Часто мышцы за счет разрастания соединительной ткани могут увеличиваться, создавая иллюзию нормальности мышц.
В дальнейшем к основным симптомам присоединяются такие признаки мышечной дистрофии: деформация скелета, аномалии развития костей, искривление позвоночника.
Осложнения мышечной дистрофии
Заболевание может поражать мышцы, связанные с дыханием. Люди с дистрофией Дюшенна редко доживают до сорока лет.
Некоторые заболевания негативно влияют на сердце, иногда возникают проблемы с приемом пищи. Потеряна способность нормально двигаться, может возникнуть паралич.
Диагностика мышечной дистрофии
Врач выясняет, болел ли кто-нибудь в семье больного подобным заболеванием, узнает о течении болезни. Он беседует с родственниками и с самим больным, оценивает его жалобы и назначает специальные исследования. Исследуются фрагменты мышечной ткани, назначается электромиография, позволяющая оценить состояние нервов в мышцах.Также проводятся иммунологические и биологические тесты.
Лечение мышечной дистрофии
Современные врачи еще не научились особым образом лечить мышечную дистрофию, чтобы полностью избавиться от болезни.
Лечение мышечной дистрофии направлено на устранение и облегчение симптомов, а также предупреждение осложнений.
Использование кортикостероидных препаратов поможет отсрочить прогрессирование дистрофии и улучшить мышечную силу.
Для тренировки и укрепления мышц назначают физические упражнения, иногда применяют гормональную и клеточную терапию.
Шины используются для предотвращения мышечных спазмов.
При различных симптомах назначают соответствующее лечение. Так, при нарушениях работы сердца применяют специальные препараты (например, фенигидин). Для облегчения ходьбы используются ортопедические приспособления. Рекомендуемые препараты, снижающие мышечную активность.
Препарат селегилин назначают при сонливости, часто сопутствующей некоторым формам заболевания.
Недавние исследования показали, что хорошие результаты дает генная терапия.Однако эта отрасль только развивается и лечение в стационарах с ней не проводится.
Альтернативное лечение мышечной дистрофии также предлагает свои методы. Вот некоторые из них.
1. Втирать масло в мышцы. После растирания больного заворачивают в простыню и оставляют полежать на час. Втирать 20 минут.
2. Скорлупу куриного яйца промыть, перемолоть в муку. В эту муку капают лимонный сок (сколько лет - столько капель), полученные комочки едят натощак и на ночь.Возьмите 20 дней.
3. Пол-литровую банку овса промывают и пересыпают в трехлитровую банку. К этому добавить чайную ложку лимонного сока, 3 большие ложки сахара и залить водой. Настоять 3 дня и пить вместо чая.
Обратитесь к неврологу!
.Мышечная дистрофия представляет собой ряд заболеваний, характеризующихся атрофией или слабостью различных мышц, наиболее распространенными из которых являются наследственные заболевания.
Существует несколько форм этого заболевания, различающихся по таким характеристикам: расположение мышц, степень атрофии или слабости, возраст начала, тип приобретения, скорость прогрессирования.
Причины заболевания точно не известны, считается, что это наследственное заболевание, связанное с нарушением обмена веществ в мышцах.Некоторые формы развиваются только у мальчиков.
Симптомы мышечной дистрофии
Основным симптомом является прогрессирующая мышечная слабость.
Дистрофия Дюшенна. Развивается чаще у мальчиков и выявляется при попытках ребенка выбраться самостоятельно. Симптомы этого вида мышечной дистрофии следующие:
Затруднение вставания из положения сидя или лежа;
Частые падения ребенка;
- походка "ладан";
Затрудненный бег и прыжки;
трудности в обучении;
Рост икроножных мышц.
Дистрофия Беккера. Симптомы этого типа мышечной дистрофии аналогичны симптомам дистрофии Дюшенна, за исключением того, что они мягче и развиваются медленнее. Первые симптомы наблюдаются в подростковом возрасте и даже после 20 лет.
Миотоническая дистрофия (болезнь Штейнерта). Благодаря ему невозможно расслабить мышцы, когда захочется. В первую очередь это касается лицевых мышц. Начинается после полового созревания.
Лопаточная дистрофия. Внешний вид человека характерен: лопатки выступают, как крылья, когда человек поднимает руки или плечи.Развивается у подростков.
Дистрофия таза и плечевой кости. Страдают мышцы плеч и бедер, отмечается невозможность поднять стопу. Развивается в раннем детстве и прогрессирует.
Врожденная дистрофия. Развивается сразу после рождения или проявляется до 2-летнего возраста. Различают легкие формы и тяжелое течение.
Глазная дистрофия. Первый признак – опущенные веки. Отмечается слабость мышц глаз, шеи, лица, затруднение глотания.Эта форма проявляется в зрелом возрасте (40-50 лет).
Часто мышцы из-за разрастания соединительной ткани могут увеличиваться, создавая иллюзию нормальной мускулатуры.
Позднее к основным симптомам присоединяются признаки мышечной дистрофии: деформация скелета, костные аномалии, искривление позвоночника.
Осложнения мышечной дистрофии
Заболевание может поражать мышцы, связанные с дыханием. Люди с дистрофией Дюшенна редко доживают до 40 лет.
Некоторые виды заболеваний отрицательно влияют на сердце, иногда возникают проблемы с приемом пищи. Потеряна способность нормально двигаться, может возникнуть паралич.
Диагностика мышечной дистрофии
Врач выясняет, болел ли кто-либо из родственников больного подобным заболеванием, узнает о течении болезни. Беседы с родственниками и самим больным, оценка его жалоб и назначение специальных обследований. Исследуются фрагменты мышечной ткани, назначается электромиография, позволяющая оценить состояние нервов в мышцах.Также проводятся иммунологические и биологические тесты.
Лечение мышечной дистрофии
Современные врачи еще не научились особым образом лечить мышечную дистрофию, чтобы полностью избавиться от болезни.
Лечение мышечной дистрофии направлено на устранение и облегчение симптомов, а также на предотвращение осложнений.
Использование кортикостероидных препаратов поможет замедлить прогрессирование дегенерации и улучшить мышечную силу.
Для тренировки и укрепления мышц назначают физические упражнения, иногда применяют гормональную и клеточную терапию.
Шины используются для предотвращения мышечных спазмов.
При различных симптомах назначают соответствующее лечение. Так, при нарушениях работы сердца применяют специальные препараты (например, фенигидин). Для поддержания походки используются ортопедические приспособления. Рекомендуются препараты, снижающие мышечную активность.
Препарат селегилин назначают при сонливости, часто сопутствующей некоторым формам заболевания.
Недавние исследования показали, что генная терапия полезна.Однако эта отрасль только развивается и лечение в стационарах с ней не проводится.
Альтернативное лечение мышечной дистрофии также предлагает свои методы. Вот некоторые из них.
1. Втирать масло в мышцы. После растирания больного заворачивают в простыню и оставляют полежать на час. Втирать нужно 20 минут.
2. Скорлупу куриного яйца промыть, перемолоть в муку. В эту муку капают лимонный сок (сколько лет - столько капель), полученные комочки едят натощак и на ночь.Возьмите 20 дней.
3. Пол-литровую банку овса промывают и пересыпают в трехлитровую банку. В него добавляют чайную ложку лимонного сока, 3 большие ложки сахара и заливают водой. Настоять 3 дня и пить вместо чая.
Обратитесь к неврологу!
Дело вот в чем Я посмотрел в электронный микроскоп и увидел (показан белым цветом на изображении) между мышечными волокнами (красный цвет).
На фото : Биопсия мышечного волокна при легкой (A), умеренной (B) и тяжелой (C) миопатии:
На фото : Нормальные мышечные волокна здорового человека:
На примере моего больного, который пострадал.Диагноз Эмине: Тяжелая мышечная дистрофия Подтверждено биопсией. Далее я опишу свой подход к устранению мышечной слабости. Рекомендую посмотреть видео о лечении прогрессирующей мышечной дистрофии Дюшенна.
Мышечная дистрофия — заболевание, при котором нарушается образование белка, входящего в состав скелета мышечной клетки.
Мышечная дистрофия начинается с развития слабости и атрофии определенной группы мышц. С годами дистрофический процесс захватывает все новые и новые группы мышц. Это происходит до тех пор, пока он полностью не обездвижен. Основным признаком миодистрофии является поражение мышц таза, плечевого пояса и туловища больного. Мышцы бедра и плеча поражаются в тяжелых случаях, как в случае пациентки Эмине: она не могла вставать без поддержки и ходить даже на короткие расстояния.
В начальном периоде, с одной стороны, может доминировать миодистрофия, но по мере прогрессирования заболевания степень поражения симметричных мышц больного становится одинаковой. Со временем течение заболевания почти во всех мышцах снижает их мышечную силу. На теле больного, страдающего мышечной дистрофией, появляются участки гипертрофированных мышц. Это псевдогипертрофия, не связанная с ростом мышечных волокон. Мышечная псевдогипертрофия связана с отеком мышц ног или рук.Эти мышцы сильные, но слабые.
Все образные формы этого заболевания у взрослых различаются:
90 134Проблемы классификации миопатий (хронических и прогрессирующих наследственных заболеваний мышц) развиваются по разным направлениям.Мышечная дистрофия у взрослых классифицируется по типу наследования:
90 102Клиническим симптомом мышечной дистрофии являются вялые параличи различных групп мышц больного без признаков поражения двигательных и периферических нервов. Неврологи весь мир не может это объяснить.
Доктор Никонова
Мой отзыв: Белковые набухания между мышечными волокнами не дают мышцам двигаться.
Незнание этого явления смущает медиков всего мира: «Как же так? Мышечное волокно интактное, неповрежденное. Двигательные нейроны и периферические нервы целы, на месте, и они прекрасно передают импульсы, идущие от мозга к мышцам и от мышц к мозгу, и движения затруднены?»
Неврологи назначают для проведения электромиографии.Опять же, для них это загадка: нет никаких изменений в структуре мышечного волокна. Снижение амплитуды М-ответа, усиление интерференции и многофазный потенциал потенциала свидетельствуют о затруднениях движения мышц без какой-либо патологии!
Давайте посмотрим, что происходит в мышечных клетках больных мышечной дистрофией Дюшенна. Для этого сделаем надрез кожи, расширим ее эспандером и возьмем небольшой кусочек мышечных волокон.
Типичным признаком миодистрофии является прежде всего разный диаметр мышечных волокон.У здорового человека диаметр мышечных волокон одинаков.
Характерными симптомами мышечной дистрофии являются атрофия и гипертрофия волокон, множественные внутренние ядра и отек.
При исследовании окрашенных фрагментов скелетных мышц я заметил денервацию миофибрилл, значительную вариабельность размеров миофибрилл и выраженный отек.
Пояснение к первой картинке :
На втором рисунке показано нормальное мышечное волокно здорового человека.
Выраженность мышечной дистрофии по данным электронной микроскопии ориентируется на следующие показатели:
На фото : Биопсия мышечного волокна при легкой (А), умеренной (В) и тяжелой (С) дистрофии.
На фото: мышечных волокна с умеренной прогрессирующей мышечной дистрофией:
(а) светло-фиолетовые мышечные волокна;
б) светлые пятна внутри мышечных волокон - припухлость, оттесняющая яички от центра клетки к периферии;
в) темные точки - ядра мышечных клеток;
г) стрелкой показана мышечная клетка, которая не может двигаться из-за замедления обменных процессов - она темнеет в сторону фиолетового цвета.
Вот состояние мышц Эмине до того, как она связалась со мной:
Пояснение к фото «Тяжелая мышечная дистрофия»:
Первым признаком миопатии Дюшенна Эмине была слабость. Она стала уставать от обычных физических нагрузок. Самые ранние жалобы Эмине:
Эмине не могла встать с низкого стула без помощи рук. При вставании женщина прибегала к вспомогательным приемам: «подъем по лестнице», «лазание в одиночку» - прием Говерса. Спустя еще несколько лет Эмине уже не могла встать из положения на корточках без посторонней помощи. Больному не разрешалось подниматься по лестнице.
После моего удара по мышцам Эмине поднимается на 17-й этаж без помощи рук, тут же спускается на лифте и без устали поднимается обратно на 17-й этаж!
Атрофия мышц развивается преимущественно в области тазового пояса, бедер (поэтому на эту область направлено эмендическое влияние на мышцы Эмине).
Позднее начинают атрофироваться мышцы верхних конечностей. Эмине сказала, что не может наливать себе чай или расчесывать волосы. Посмотрите результаты лечения мышечной дистрофии в видео ниже:
Отличаются избирательным распространением слабостей и специфичностью генетических аномалий.
Дистрофия Беккера, хотя и тесно связанная с ней, имеет более позднее начало и вызывает более легкие симптомы. Другие формы включают дистрофию Эмери-Дрейфуса, миотоническую дистрофию, поясную дистрофию, лицевую лопаточно-плечевую дистрофию и врожденную дистрофию.
Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера представляют собой Х-сцепленные рецессивные заболевания, характеризующиеся прогрессирующей слабостью проксимальных мышц из-за дегенерации мышечных волокон. Дистрофия Беккера имеет позднее начало и вызывает более легкие симптомы. Лечение направлено на поддержание функции с помощью физиотерапии, ортопедических приспособлений и ортопедических приспособлений; преднизолон назначают некоторым пациентам с тяжелыми функциональными нарушениями.
При дистрофии Дюшенна эта мутация вызывает тяжелое отсутствие (
Дистрофия Дюшенна. Расстройство обычно появляется в возрасте от 2 до 3 лет. Слабость поражает проксимальные мышцы, обычно в первую очередь нижние конечности. Дети часто ходят на цыпочках, имеют утиную походку и лордоз. Нарастание слабости стабильное, развиваются сгибательные контрактуры конечностей, сколиоз. Развивается достоверная псевдогипертрофия. Большинство детей прикованы к инвалидной коляске к 12 годам.Заболевания сердца обычно протекают бессимптомно, хотя у 90% пациентов наблюдаются отклонения на ЭКГ. Треть из них имеют легкие непрогрессирующие умственные нарушения, которые больше влияют на вербальные навыки, чем на производительность.
Дистрофия Беккера. Это расстройство обычно проявляется симптоматически гораздо позже и легче. Способность ходить обычно сохраняется до 15 лет и старше, и многие дети остаются подвижными и во взрослом возрасте. Большинство жертв доживает до 30-40 лет.
Диагноз предполагается на основании клинических признаков, возраста начала заболевания и семейного анамнеза, предполагающего рецессивный тип наследования, сцепленный с Х. Миопатические изменения выявляются с помощью электромиографии (потенциалы двигательных единиц быстро возрастают, короткие по продолжительности и с низкой амплитудой) и при биопсии мышц (некроз и заметное изменение размеров мышечных волокон, не отделившихся от двигательных единиц).Уровни креатинкиназы в 100 раз выше нормы.
Диагноз подтверждается тестом на дистрофин с иммуноокрашиванием биопсии. Дистрофии у больных дистрофией Дюшенна не выявляют. Анализ мутаций ДНК в лейкоцитах периферической крови также может подтвердить диагноз, если выявляется аномалия в гене дистрофина (примерно у 65% больных имеются делеции или дупликации, примерно у 25% больных — точечные мутации).
Специфического лечения нет. Умеренные физические нагрузки рекомендуются как можно дольше. Ортез на лодыжку поможет предотвратить сгибание во время сна. Ортезы для ног могут помочь вам временно сохранить способность стоять и ходить. Следует избегать ожирения; потребность в калориях, вероятно, будет ниже, чем обычно. Показано генетическое консультирование.
Ежедневный прием преднизолона не приводит к значительному долгосрочному клиническому улучшению, но может замедлить течение заболевания.Нет единого мнения о долгосрочной эффективности. Генная терапия еще не разработана. Иногда необходима корректирующая операция. Респираторные расстройства иногда можно лечить с помощью неинвазивной респираторной поддержки (через носовую маску). Набирает популярность плановая трахеотомия, позволяющая детям с дистрофией Дюшенна дожить до 20 лет.
Дистрофия Эмери-Дрейфуса ... Это заболевание может наследоваться по аутосомно-доминантному, аутосомно-рецессивному (наименее часто) или сцепленному с Х-хромосомой типу.Общая частота неизвестна. Носителями могут быть самки, но только самцы клинически страдают Х-сцепленным наследованием Гены, связанные с дистрофией Эмери-Дрейфуса, кодируют белки ядерной мембраны, А/С (аутосомно) ламину и эмерин (Х-сцепленный).
Мышечная слабость и истощение могут возникнуть в любое время до 20 лет и обычно связаны с бицепсами, трицепсами и, реже, с дистальными мышцами ног. Сердце часто ассоциируется с мерцательной аритмией, нарушением проводимости (АВ-блокада), кардиомиопатией и высокой вероятностью внезапной смерти.
Диагноз устанавливается на основании клинических результатов, возраста начала заболевания и семейного анамнеза. А также слегка повышенный уровень креатинкиназы в сыворотке и миопатические результаты электромиографии и биопсии мышц. Диагноз подтверждается тестами ДНК.
Лечение включает противоконтрактурную терапию. Кардиостимуляторы иногда необходимы пациентам с нарушениями проводимости.
Миотоническая дистрофия ... Миотоническая дистрофия является наиболее распространенной формой мышечной дистрофии у белого населения.Это происходит примерно у 30/100 000 живорожденных мужчин и женщин. Наследование аутосомно-доминантное с переменной пенетрантностью. Два генетических локуса - DM 1 и DM 2 - вызывают аномалии. Симптомы и признаки появляются в подростковом или юношеском возрасте и включают миотонию (замедленное расслабление после мышечного сокращения), слабость и истощение дистальных мышц конечностей (особенно рук) и лицевых мышц (особенно птоз) и кардиомиопатию. Также могут развиться умственная отсталость, катаракта, эндокринные нарушения.
Диагноз подтверждается характерными клиническими симптомами, возрастом начала заболевания и семейным анамнезом; диагноз подтверждается тестами ДНК. Лечение включает использование ортопедического аппарата при дряблости стопы и медикаментозную терапию миотонии (например, мексилетин по 75-150 мг внутрь 2-3 раза в день).
Поясно-конечностная дистрофия ... В настоящее время известен 21 подтип поясно-конечностной дистрофии: 15 аутосомно-рецессивных и 6 аутосомно-доминантных. Общая частота неизвестна.Было идентифицировано несколько хромосомных локусов для аутосомно-доминантных (5q [продукт гена неизвестен)) и рецессивных (2q, 4q [, 13q [γ-саркогликан]], 15Q [кальпаин, Са-активируемые протеазы] и 17q [α-саркогликан или адхалин ]) формы . Могут быть затронуты структурные (например, гликопротеины, родственные дистрофину) или неструктурные (например, протеазы) белки.
Симптомы включают слабость в поясничном отделе и проксимальных отделах конечностей. Начало заболевания колеблется от раннего детства до зрелого возраста; начало аугосомно-рецессивных типов, как правило, в детском возрасте, и эти типы в основном связаны с поражением тазового пояса.
Диагноз подтверждается характерными клиническими симптомами, возрастом начала заболевания и семейным анамнезом; диагностика также требует определения гистологической картины мышц, иммуноцитохимии, вестерн-блоттинга и генетических тестов на наличие специфических белков.
Лечение направлено на профилактику контрактур.
Фазно-лопаточно-плечевая дистрофия ... Начало заболевания в подростковом или подростковом возрасте характеризуется медленным прогрессированием: ребенку трудно свистеть, закрывать глаза и поднимать руки (из-за ослабления мышцы, стабилизирующие лопатку).Продолжительность жизни нормальная. Детские разновидности, характеризующиеся слабостью в области лица, плеч и тазового пояса, быстро прогрессируют.
Диагноз подтверждается характерными клиническими симптомами, возрастом начала заболевания и семейным анамнезом; диагноз подтверждается анализом ДНК.
Лечение состоит из физиотерапии.
Врожденная мышечная дистрофия ... Это не заболевание само по себе, это врожденный дефект, относящийся к одной из немногих редких форм мышечной дистрофии.Диагноз подозревается у любого летаргического новорожденного, но его следует отличать от врожденной миопатии с помощью биопсии мышц.
Лечение — это физиотерапия, которая помогает поддерживать функцию мышц.
Существует множество форм мышечной дистрофии. Они различаются по таким характеристикам, как возраст начала заболевания, локализация пораженных мышц, выраженность мышечной слабости, скорость прогрессирования дистрофии, тип наследования. Наиболее распространены две формы: мышечная дистрофия Дюшенна и миотоническая мышечная дистрофия.
(псевдогипертрофическая мышечная дистрофия) — наиболее частая форма заболевания у детей. Заболевание вызывается генетическим дефектом, локализованным в Х-хромосоме (одна из двух хромосом, определяющих пол человека). Женщины с дефектным геном передают его своим детям, но сами обычно не имеют симптомов дистрофии. У мальчиков с дефектным геном неизбежно развивается мышечная слабость в возрасте от двух до пяти лет.
В первую очередь это касается крупных мышц нижних конечностей и тазового пояса.Затем дегенерация распространяется на мышцы верхней части тела, а затем постепенно на все основные группы мышц. Характерным признаком заболевания является псевдогипертрофия икроножной мышцы, т.е. ее увеличение в результате отложения жира и разрастания соединительной ткани. Однако при истинной мышечной гипертрофии увеличивается объем самой мышечной ткани.
Мышечная дистрофия Дюшенна — одна из наиболее тяжелых и быстро прогрессирующих форм. К 12 годам больные обычно теряют способность двигаться, а к 20 годам большинство из них погибает.
(болезнь Штейнерта) — наиболее распространенная форма мышечной дистрофии у взрослых. Это происходит из-за дефектного гена на хромосоме 19. Мужчины и женщины страдают в равной степени и могут передать генетический дефект своим детям. Заболевание проявляется в любом возрасте, включая младенческий, но чаще всего в возрасте от 20 до 40 лет. Первыми симптомами являются миотония (замедленное расслабление мышц после сокращения) и слабость мышц лица; также возможно поражение мышц конечностей и других частей тела.Заболевание в большинстве случаев прогрессирует медленно, а полная инвалидизация может наступить не раньше, чем через 15 лет.
Особенность этого заболевания в том, что помимо произвольных мышц поражаются также гладкие мышцы и сердечная мышца.
Все формы мышечной дистрофии характеризуются дегенерацией мышц, но не связанных с ними нервов. В пораженной мышечной ткани обнаруживают различные изменения, в том числе значительные колебания толщины (диаметра) мышечных волокон.Постепенно эти волокна теряют способность сокращаться, распадаются и замещаются жировой и соединительной тканью.
По своим клиническим проявлениям мышечные дистрофии сходны со спинальными амиотрофиями — наследственным заболеванием, поражающим двигательные нейроны спинного мозга. Эти заболевания также приводят к выраженной мышечной слабости, иногда опасной для жизни. Для подтверждения диагноза мышечной дистрофии может потребоваться электромиография, а иногда и биопсия мышц с микроскопическим исследованием для выявления характерных дистрофических изменений.
Специалисты считают, что каждая форма мышечной дистрофии вызвана определенным точечным генетическим дефектом, который препятствует способности мышечных клеток синтезировать необходимые белки. Усилия исследователей сосредоточены на поиске основных дефектов болезни и белковых аномалий, к которым эти дефекты приводят. Выявлен ген мышечной дистрофии Дюшенна.
Невозможно предотвратить или замедлить прогрессирование мышечной слабости при мышечной дистрофии.Терапия в основном направлена на борьбу с такими осложнениями, как деформация позвоночника, развивающаяся из-за слабости мышц спины, или склонность к пневмонии из-за ослабления дыхательной мускулатуры. В этом направлении достигнуты определенные успехи и улучшение качества жизни больных мышечной дистрофией. Многие пациенты сегодня, несмотря на свое заболевание, могут вести полноценную и продуктивную жизнь.
Болезнь структур связочного аппарата - Мышечная дистрофия - объединяет несколько форм патологии, некоторые из которых протекают тяжело и угрожают жизни.
90 100Патология затрагивает глубокие слои мягких тканей, чаще всего относится к прогрессирующим формам. Это означает, что однажды выявленное заболевание будет продолжать развиваться, уменьшая прочность мышечной ткани и диаметр волокон.
Прогрессирующая мышечная дистрофия неизбежно приводит к полному расщеплению некоторых волокон, но болезнь можно замедлить, предотвратив быструю дегенерацию тканей. По мере развития заболевания мышечный участок постепенно замещается слоем жира.
Ученые не могут найти точных причин заболевания, но выделяют мутации, ответственные за развитие патологии. Таким образом, в 100% случаев происходит изменение аутосомно-доминантного гена, отвечающего за выработку белка, участвующего в формировании и поддержании мышечных волокон.
Поврежденная хромосома указывает на место развития мышечной дистрофии:
Как наследственные, так и приобретенные формы мышечной дистрофии могут вызывать патологические изменения.
Независимо от того, какой вид мышечной дистрофии у больного имеется или начинает развиваться, все они имеют общий набор симптомов:

По мере прогрессирования мышечной дистрофии появляются дополнительные симптомы, каждый из которых соответствует определенному виду патологии.
Большинство мышечных дистрофий относятся к категории врожденных патологий, связанных с аномалиями генома. Однако существуют формы, при которых мутации возникают в результате воздействия токсических веществ.
Псевдогипертрофия прогрессирующего типа.Находят даже в детском возрасте, потому что симптомы ярко выражены и быстро ухудшаются. Почти все заболевшие мужчины, но есть больные и среди девушек.
Симптомы мышечной дистрофии достаточно выражены в 2-летнем возрасте, к 5 годам достигают пика:
Эта прогрессирующая мышечная дистрофия приводит к смерти в 100% случаев в возрасте до 30 лет. Большинство из них моложе 20 лет.
Мышечная Дистрофия Штейнерта развивается у взрослых в возрасте от 20 до 40 лет и характеризуется поздними симптомами. В редких случаях патология возникает в детском возрасте. Врачи не замечают особой зависимости от пола.Он течет медленно, его можно сдерживать.
90 100Важно! Отличительной чертой нарушения является то, что процесс проникает в структуры жизненно важных органов, провоцируя слабость мышц лица и других областей.
Пищевые волокна расщепляются медленно, но заболевание требует постоянного наблюдения врачей. Если мышечная дистрофия поражает легкие или сердце, смерть может наступить быстро.
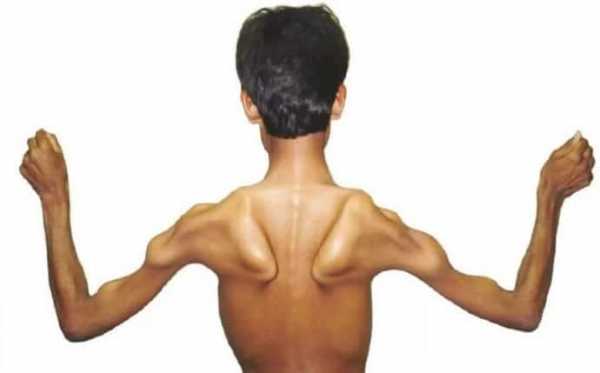
Синдром Беккера относится к прогрессирующим мышечным дистрофиям.Встречается довольно редко и развивается медленно. Чаще всего заболевание возникает у людей невысокого роста. Заболевание поддается лечению и легко контролируется, удается затормозить патологический процесс на 20-30 лет при сохранении стандартной эффективности. Инвалидность наступает только при наличии дополнительных заболеваний или тяжелых травм.
Первые симптомы мышечной дистрофии появляются в период от 10 до 20 лет.Заболевание развивается медленно, начиная с плечевого пояса и рук, затем отступают и другие мышцы. У человека прогрессирующие мышечные дистрофии вызывают резкое изменение осанки – грудная клетка смещается назад, а живот резко выпячивается вперед. Врачи описывают симптомы как «утиную прогулку».
Симптомы мышечной дистрофии впервые появляются у детей с 6 лет, но могут появиться и до 52 лет.Чаще всего первые признаки регистрируются между 10 и 15 годами. В первую очередь поражаются лицевые мышцы, затем конечности и крупные мышцы туловища.
90 100Важно! Первым признаком расстройства является неполное смыкание век во время сна. Затем рот перестает закрываться как в состоянии покоя, так и в состоянии бодрствования, что сильно влияет на дикцию.
Мышечная дистрофия развивается медленно, длительное время больной сохраняет нормальную двигательную активность, может заниматься привычными делами.Атрофия тазового пояса, приводящая к инвалидности, развивается в основном через 20-25 лет после выявления патологии. При правильном лечении заболевание длительное время не проявляется комплексными симптомами.

Этот вид мышечной дистрофии не имеет ничего общего с геномом человека, так как развивается только на фоне длительного употребления высоких доз алкоголя. Сопровождается сильными болями в конечностях из-за разрыва мышечных волокон. Хроническая миопатия протекает с умеренными симптомами, а острая миопатия проявляется воспалением и приступами боли.

Дистальная мышечная дистрофия — легкое состояние, которое трудно выявить из-за отсутствия тяжелых симптомов. Диагноз часто путают с нервной амиатрофией Мари-Шарко. Для дифференциальных исследований требуется головная энцефалограмма. Общие симптомы заболевания очень похожи на многие другие патологии.

Специфических методов диагностики этой формы заболевания нет, она очень похожа на синдром Дюшенна, но есть специфические симптомы.Они появляются достаточно редко, так как сам синдром встречается гораздо реже, чем другие виды мышечной дистрофии.
В большинстве случаев патология развивается в возрасте до 30 лет, при этом поражаются мышцы сердца. Патология характеризуется наличием порока сердца, но проявляется слабо выраженными симптомами. Если не исправить, это может привести к смерти.
При этой форме мышечной дистрофии страдают нервные связи, отвечающие за двигательную активность.В результате изменяются мышцы спинного мозга и более глубоких тканей. Нарушается структура ядер нервных клеток, в первую очередь страдают мимические мышцы и глаза.
Патология имеет множество симптомов, некоторые из которых поражают сенсорные рецепторы: ощущения могут усиливаться или ослабевать. Иногда возникают головокружение, судороги, болезни сердца и проблемы со зрением. Поражение потовых желез.
Заболевание связано с наследственными нарушениями.Сначала вовлекаются мышцы поясницы и туловища, затем верхних конечностей. Мышцы лица почти никогда не вовлекаются в заболевание. Заболевание прогрессирует медленно, легко контролируется медикаментозно и не приводит к быстрой инвалидизации.
Этот вид мышечной дистрофии характеризуется поздними симптомами и возникает во взрослом возрасте. Чаще всего страдают определенные этнические группы. Симптомы появляются в возрасте 25-30 лет:
Постепенно в процесс могут вовлекаться и другие мышцы черепа, но это бывает не всегда. В некоторых случаях поражаются мышцы плечевого пояса и шеи. Из-за этого возникают проблемы с речью и дикцией.
Прогрессирующая мышечная дистрофия у детей протекает по-разному и более опасна осложнениями, чем первичная атрофия мышц. Даже незначительная инфекция или патология дыхательной системы могут привести к летальному исходу из-за быстрого развития и вовлечения других органов.Иногда заподозрить мышечную дистрофию слишком сложно, родители должны насторожиться при появлении симптомов:
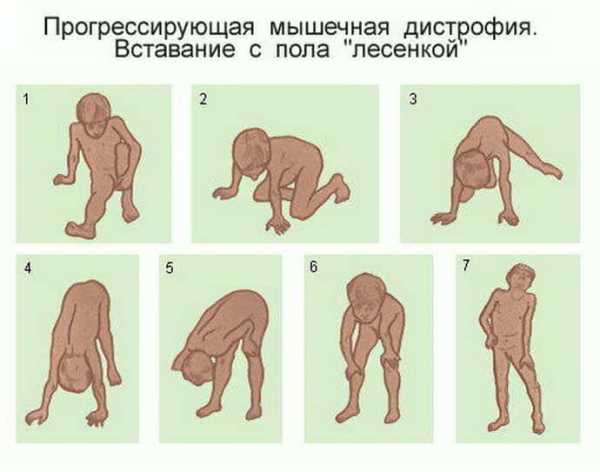
Формы заболевания могут иметь разные названия, но большинство из них имеют схожие симптомы.
Диагностировать мышечную дистрофию можно после прохождения клинического исследования:
При диагностике необходимо указать название белка, синтез которого недостаточен в организме.
Лечение мышечной дистрофии необходимо начинать с устранения опасных симптомов, так как реальных методов коррекции для решения генетических проблем до сих пор не существует. Например, при поражении спинных мышц назначают препараты для повышения тонуса.
90 100Важно! Если патология опасна осложнениями со стороны сердечной системы, иногда имплантируют кардиорегулятор.
Большинство препаратов относятся к группе сильнодействующих и назначаются врачом строго по назначению врача.Кроме медикаментов врачи рекомендуют использовать ортопедические приспособления для укрепления мышц и рук. Они также используют анаболический стероид для укрепления мышечной ткани.

Генная терапия сложна и ненадежна, но она быстро развивается. Например, при лечении синдрома Дюшенна используется искусственно созданный ген, который затем имплантируется человеку. Для этого нужный ген встраивают в аденовирус и вводят в мышечную ткань.
Чаще всего мышечная дистрофия приводит к развитию жизнеугрожающих осложнений: легочных и сердечных нарушений, снижению двигательной активности и параличу, искривлению позвоночника и ухудшению интеллектуальных способностей.
Диагноз мышечная дистрофия может быть поставлен пациенту как смертный приговор, но в долгосрочной перспективе. Патология легче всего протекает у взрослых. Если заболевание обнаружено у ребенка, шанс, что он доживет до 20 лет, катастрофически мал.Однако поддерживающая терапия может продлить активную жизнь пациента и свести к минимуму риск осложнений.
.Генетические заболевания относятся к тяжелым патологиям, лечение которых на сегодняшний день затруднено. Среди сходных хромосомных аномалий встречаются различные нарушения. Многие из них имеют неврологическую симптоматику. Примерами являются миодистрофия Дюшенна, Беккера. Эти заболевания развиваются в детском возрасте и прогрессируют. Несмотря на достижения неврологии, такие патологии не поддаются лечению. Это связано с хромосомными изменениями, происходящими в процессе формирования организма.
Миодистрофия Дюшенна представляет собой генетическое заболевание, проявляющееся прогрессирующим нарушением мышечной системы. Патология встречается редко. Частота аномалии составляет около 3 человек на 10 000 мужчин. Болезнь поражает мальчиков почти во всех случаях. Тем не менее не исключено развитие миодистрофии у девочек. Данная патология проявляется в раннем детстве.
Другим заболеванием, имеющим те же причины и симптомы, является миодистрофия Беккера.Отличается более благоприятным течением. Поражение мышечной ткани происходит значительно позже – в подростковом возрасте. В этом случае симптомы развиваются постепенно, и больной сохраняет трудоспособность в течение нескольких лет. Как и при миодистрофии Дюшенна, эта патология распространена среди мужского населения. Частота встречаемости – 1 случай на 20 тысяч мальчиков.
Причиной обеих патологий является нарушение Х-хромосомы.Генетические изменения при инфарктах миокарда Беккера и Дюшенна изучались в 1930-х годах. Тем не менее, этиологическая терапия до сих пор не найдена. Тип наследования аномалии рецессивный. Это означает, что при наличии патологического гена у одного из родителей вероятность рождения больного ребенка составляет 25%. Х-хромосома самая длинная в организме. При обоих типах дистрофии заболевание возникает в одном и том же локусе (p21). Эти повреждения приводят к снижению синтеза белка, входящего в состав клеточных мембран мышечной ткани.При миодистрофии Дюшенна она полностью отсутствует. Поэтому нарушения возникают гораздо раньше. При миодистрофии Беккера белок синтезируется в малых количествах или является патологическим.
Миодистрофия Дюшенна характеризуется поражением нервно-мышечного аппарата. Болезнь можно заподозрить в 2-3-летнем возрасте. В этот период выясняется, что ребенок отстает в физическом развитии от своих сверстников, плохо ходит, бегает и прыгает. Этим детям трудно подниматься по лестнице, она часто падает.Поражение мышц начинается с нижних конечностей. Позже она распространяется на все проксимальные отделы мышц. Дегенерация происходит в верхней плечевой окружности, четырехглавой мышце бедра. В этих местах происходит истончение мышц. Миодистрофия развивается с годами. Поражение мышц и постоянное давление на них приводят к контрактурам — непрерывному искривлению конечностей. Кроме того, у больных с миодистрофией Дюшенна периодически проявляется порок сердца. Также для данной патологии характерно снижение интеллектуальных способностей (не сильно выраженное).
Мышечная дистрофия Беккера имеет те же симптомы, но развивается позднее. Первые симптомы наблюдаются в 10-15 лет. Наблюдается постепенное изменение походки, появляется тремор, позже развиваются контрактуры. Нарушения со стороны сердечно-сосудистой системы легкие. Интеллект при этом заболевании обычно не снижается.
Диагноз «миодистрофия Дюшенна» (или Беккера) может поставить опытный невролог. Прежде всего, он основан на клинической картине этих заболеваний.Обращают на себя внимание такие симптомы, как истончение проксимальных мышц, ложная гипертрофия икроножных мышц (обусловленная фиброзом и отложением жира). Эти симптомы почти всегда связаны с сердечно-сосудистыми заболеваниями. На ЭКГ можно заметить нарушение ритма, гипертрофию левого желудочка.
Также больные миодистрофией Дюшенна немного отстают от своих сверстников в умственном развитии. Чтобы определить это, с детьми работает психолог. При подозрении на это заболевание проводят миографию (определение электрического потенциала мышц) и ЭхоКС - исследование камер сердца.Для точного определения наличия патологии проводят генетическую диагностику. При мышечной дистрофии Бедыря и Дюшенна больные должны находиться под наблюдением нескольких специалистов. Среди них невролог, психолог и кардиолог.
К сожалению, этиологические методы лечения миодистрофии по Китчену и Беккеру не разработаны. Тем не менее, пациентам показана симптоматическая и поддерживающая терапия. На ранних стадиях заболевания предлагаются курсы лечебной физкультуры и массажа.При значительной степени инвалидности необходимо совершать пассивные движения конечностями. Для замедления прогрессирования развития разгибательных контрактур прибегают к фиксации ног во время сна. Поддерживающая терапия может продлить жизнь пациентов и уменьшить симптомы заболевания. Применяются препараты кальция, препараты «Галантамин» и «Прозерин». В некоторых случаях приходится назначать гормональные средства, преимущественно «преднизолон». В случае прогрессирующих заболеваний сердца рекомендуется применение кардиопротекторов.
Прогноз при миодистрофии Дюшенна неутешительный.Раннее развитие симптомов и быстрое прогрессирование заболевания приводят к детской инвалидности. Больные с данной патологией нуждаются в постоянном уходе. Средняя продолжительность жизни больных составляет около 20 лет. Мышечная дистрофия Беккера имеет благоприятное течение. При постоянном наблюдении врачей и выполнении их указаний трудоспособность больных сохраняется до 30-35 лет.
.